
Abstract
To determine the range of neurodevelopmental diagnoses associated with intermediate (45-54 repeats) and premutation (55-200 repeats) range cytosine-guanine-guanine fragile X expansions, the medical records of children with intermediate or premutation range expansions were retrospectively reviewed, and all neurodevelopmental diagnoses were abstracted. Twenty-nine children (9 female, 20 male; age, 13 months to 17 years) with intermediate (n = 25) or premutation (n = 4) range expansions were identified with neurodevelopmental diagnoses, including global developmental delay/intellectual disability (n = 15), language and learning disorders (n = 9), attention-deficit hyperactivity disorder (n = 5), epilepsy (n = 5), and motor disorders (n = 12), including 2 boys younger than 4 years of age with tremor and ataxia. Thus, children with intermediate or premutation range fragile X cytosine-guanine-guanine expansions may be more susceptible than children without such expansions to other processes, both genetic and environmental, that contribute to neurodevelopmental disability.
Fragile X syndrome is the most prevalent cause of heritable intellectual disability. 1 The fragile X mental retardation 1 gene facilitates brain development and dendrite maturation. 2,3 In the general population, the fragile X mental retardation 1 gene comprises a limited number of cytosine-guanine-guanine repeats. Individuals with cytosine-guanine-guanine expansions of more than 200 repeats are considered to have a full mutation and are diagnosed with fragile X syndrome. Individuals with fragile X syndrome typically demonstrate mild to severe intellectual disabilities and behavioral impairments, ranging from attention-deficit hyperactivity disorder (ADHD) to autism spectrum disorders. Individuals with expansions of 5 to 44 cytosine-guanine-guanine repeats are considered to be unaffected by fragile X syndrome. However, individuals with expansions of 45 to 54 repeats are considered to be in an intermediate range, and those with expansions of 55 to 200 repeats are considered premutation carriers for fragile X syndrome. 3 When a premutation allele is inherited from mother to child, the number of repeats may expand, resulting in fragile X syndrome. This expansion may result in deleterious effects in 2 possible ways. 3 First, production of the fragile X mental retardation protein will be reduced or absent when the cytosine-guanine-guanine expansion is too great. Without the fragile X mental retardation protein to act as a braking system on the production of proteins associated with learning and memory, the brain creates too many synaptic connections that may not be fully complete or functional. 4 Second, there is evidence that cytosine-guanine-guanine repeats in the premutation range result in increased levels of fragile X mental retardation 1 messenger ribonucleic acid (RNA), which has toxic effects on human neural cell lines. 3,5,6
While possessing the propensity for meiotic instability that results in an increased risk for fragile X syndrome in future generations, those with intermediate and premutation range cytosine-guanine-guanine repeats have historically been considered to be free from neurodevelopmental sequelae. However, recent evidence has been emerging that both younger (ADHD, autism spectrum disorders) 1,7,8 and older (fragile X–associated tremor/ataxia syndrome) 5,9 premutation carriers may in fact demonstrate neurological symptoms caused by the premutation. 10 –12 Given these recent reports of neurologically symptomatic premutation carriers, our objective for this study was to determine the range of potential neurodevelopmental disability associated with these historically asymptomatic cytosine-guanine-guanine expansion sizes.
Methods
We retrospectively reviewed the electronic medical records of children and adolescents from birth to 18 years of age who were evaluated at the Mayo Clinic in Rochester, Minnesota, from 1996 to 2008 and who were identified to have intermediate or premutation range cytosine-guanine-guanine expansions based on a search of our laboratory database. All information related to neurological concerns, neurodevelopmental diagnoses, and laboratory work-up was abstracted from their medical records. Only children whose families provided research authorization for medical record review were included in this study. This study was approved by the Institutional Review Board of the Mayo Clinic. These cases represent a clinical population because they were seen at the Mayo Clinic for clinical concerns and not population screening.
Results
There were 1447 deoxyribonucleic acid (DNA) tests for fragile X mental retardation 1 mutations performed for individuals aged 18 years or younger at our institution during the study period. Of these, 5 were identified as having a premutation range repeat size, 4 of whom provided research authorization for medical record review. All 4 of these children were found to have at least 1 neurodevelopmental diagnosis. Twenty-eight individuals were identified as having an intermediate range repeat size. Of these, 25 provided research authorization for medical record review, and all 25 were found to have at least 1 neurodevelopmental diagnosis. Table 1 summarizes the patient characteristics. Those with intermediate range repeat sizes represented 86% of our sample. While there was an equal number of males and females in the premutation group, males outnumbered females 2.5:1 in the intermediate group.
Patient Characteristics
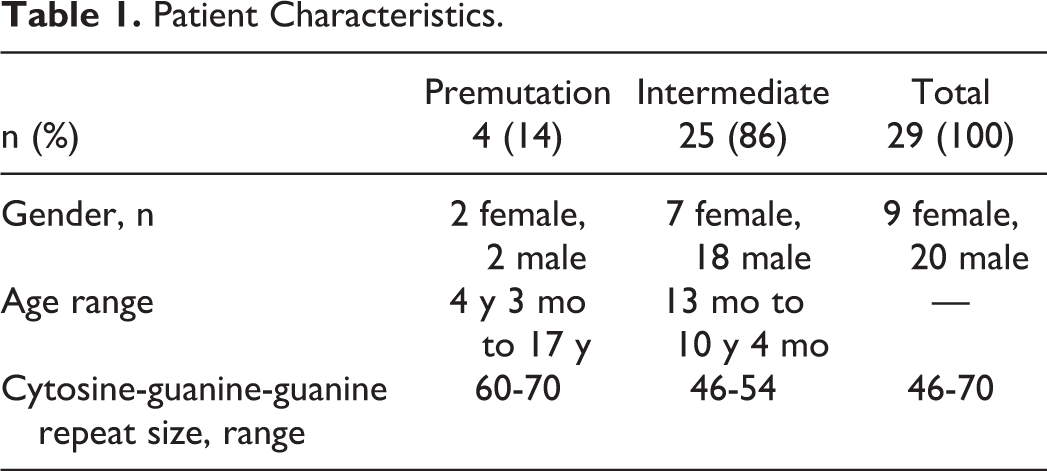
Table 2 summarizes the neurodevelopmental diagnoses found in both the premutation and intermediate groups. Overall, 52% of our patients were found to have global developmental delays or intellectual disabilities, 31% were found to have language or learning disorders, 10% were found to have autism spectrum disorders (1 female with a premutation was found to have Asperger syndrome, and 2 males with intermediate range repeat sizes were found to have pervasive developmental disorder, not otherwise specified), 17% were found to have ADHD, 17% were found to have epilepsy, and 41% were found to have some type of motor disorder, including 4 with tremors, 3 with hypotonia, 3 with developmental dyspraxia, 3 with motor tics, and 2 with ataxia. There were 66% of children in this study who had more than 1 neurodevelopmental diagnosis. No patient was found to have any other identifiable genetic or metabolic disorder, toxic environmental exposure, or structural brain abnormality to account for his or her neurodevelopmental diagnosis.
Children With Premutation and Intermediate Range Fragile X Cytosine-Guanine-Guanine Expansions With Each Neurodevelopmental Diagnosis
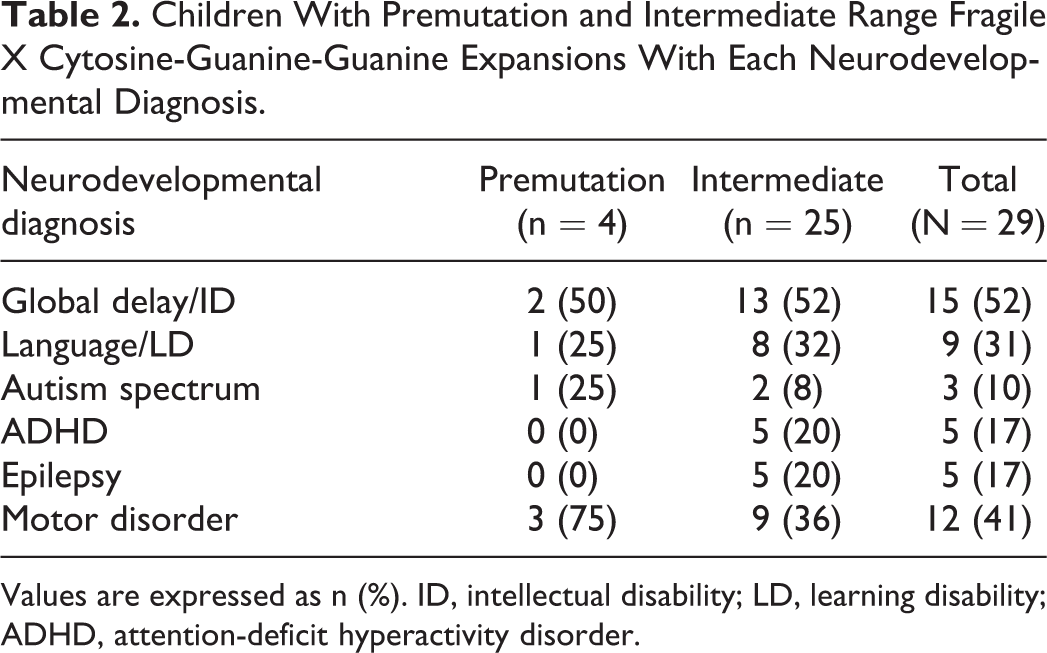
Values are expressed as n (%). ID, intellectual disability; LD, learning disability; ADHD, attention-deficit hyperactivity disorder.
Our medical record review identified 2 boys younger than 4 years of age, one with an intermediate range of 48 repeats and one with a premutation range of 64 repeats, with both tremor and ataxia, possibly representing a pediatric equivalent of fragile X–associated tremor/ataxia syndrome (FXTAS), although the radiological features of fragile X–associated tremor/ataxia syndrome in older carriers were not present, and they did not have progression of their symptoms as is seen in older carriers.
Discussion
The importance of identifying premutation carriers of fragile X syndrome is evolving, as more evidence is accumulating that suggests that premutation carriers may be at risk for a wide range of neurodevelopmental disorders. 1,10,13 Historically, fragile X premutation carriers had been considered to be asymptomatic, although primary ovarian insufficiency and emotional problems have been reported for many years. 14 Identifying premutation carriers was only deemed to be important because of the carriers’ capacity for the transmission of fragile X syndrome to future generations. More recent literature, however, has demonstrated that premutation carriers are at increased risk for a range of neurological, psychiatric, endocrine, and autoimmune disorders across their life span. 10,15 –19 Fragile X–associated tremor/ataxia syndrome has now been identified as being related to the expansion of cytosine-guanine-guanine repeats in the premutation range in older adult males and females. Fragile X–associated tremor/ataxia syndrome usually occurs in older men who exhibit late-onset ataxia with an action tremor. Other associated features of fragile X–associated tremor/ataxia syndrome include neuropathy, memory loss, executive functioning deficits, cognitive decline, muscle weakness, psychiatric problems, and autonomic dysfunction. 20,21 The magnetic resonance imaging (MRI) features of fragile X–associated tremor/ataxia syndrome include brain atrophy, white matter disease, and usually involvement of the middle cerebellar peduncles. 22 Neuropathological features include eosinophilic intranuclear inclusions in neurons and astrocytes throughout the brain and in the peripheral nervous system. 12,23 In addition to fragile X–associated tremor/ataxia syndrome, more recent studies demonstrate that premutation carriers may exhibit other difficulties such as hypertension, fibromyalgia, and hypothyroidism. 10 In addition, there is a neurodevelopmental component to premutation involvement, leading to problems such as social deficits, autism spectrum disorders, and attention problems. 1,7,8 In a clinical sample of probands identified with the premutation, there was a significantly higher rate of autism spectrum disorders and ADHD in young male premutation carriers compared to their brothers without the premutation. 1
This new evidence suggests that premutation carriers may be at risk for a wide range of neurological sequelae. In this case series, we have identified a range of neurodevelopmental disorders (Table 2) among children with cytosine-guanine-guanine repeats in an intermediate and premutation range. While studies of premutation carriers have shown that the prevalence of fragile X–associated tremor/ataxia syndrome increases with age (17% of male carriers are in their 50s, 38% in their 60s, 47% in their 70s, and 75% in their 80s), 9 in this case series, We have identified 2 children with tremor and ataxia (one with the premutation and one with an intermediate allele), suggesting that these types of motor problems may also be related to pediatric involvement from these alleles.
While it is certainly possible that each of the children included in our case series may have another as of yet unidentified cause for their neurodevelopmental disorders, at this point, despite aggressive genetic and metabolic work-ups, no alternative diagnoses have been made. It has been shown that an expanded cytosine-guanine-guanine repeat results in increased levels of fragile X mental retardation 1 messenger RNA, which has toxic effects on neural cell lines, 6,20,24 so it is likely that the premutation can cause developmental problems in some carriers, particularly those who may experience additional genetic or environmental toxicity. It has been shown that in the neuronal cell cultures of the premutation, there is earlier cell death of premutation neurons compared to controls. 6 We hypothesize that children with the premutation may be more susceptible than children without the premutation to other processes, both genetic and environmental, that contribute to neurodevelopmental disability. 3,25 Thus, it is certainly possible that individuals with premutations may be more vulnerable to secondary insults that might not necessarily result in neurodevelopmental disability in the general population. Due to their expanded cytosine-guanine-guanine repeat sizes and resultant messenger RNA toxicity, a secondary insult may be more likely to result in deleterious effects on brain development and subsequent neurodevelopmental disability in those with premutations. Given the range of neurodevelopmental disability found in our patients with both intermediate range and premutation range cytosine-guanine-guanine repeats, measurement of messenger RNA levels may become important for all individuals with cytosine-guanine-guanine expansions, especially to determine if intermediate range cytosine-guanine-guanine repeats can truly be associated with a disability. 13 Previous reports have demonstrated an increase in fragile X mental retardation 1 messenger RNA in the intermediate or gray zone alleles, 26 and there is a higher rate of premature ovarian insufficiency in women with the intermediate repeat compared to the general population. 27,28 Unfortunately, we do not have messenger RNA levels to report for any patient in our case series.
An expanded cytosine-guanine-guanine repeat, as demonstrated by individuals with fragile X–associated tremor/ataxia syndrome, can also be associated with specific findings on brain MRI scans. 3,5,9 The MRI findings include white matter disease in the subcortical, middle cerebellar peduncle, and periventricular regions. Approximately 60% of adults with fragile X–associated tremor/ataxia syndrome have been noted to have focal abnormal T2 hyperintensity in the cerebellum. Currently, abnormal T2 hyperintensity in the middle cerebellar peduncle region is used as a supporting diagnostic factor for fragile X–associated tremor/ataxia syndrome. 5 However, no abnormal signal intensity within the posterior fossa was seen in our pediatric patients with tremor and ataxia.
In association with other recent reports, this case series highlights the need to potentially consider intermediate and premutation range fragile X carriers in a new light. DNA testing for fragile X syndrome should be a routine component of the laboratory work-up of children presenting with developmental delays or autism spectrum disorders. 29,30 Our findings indicate that this testing could also be indicated for children presenting with hypotonia, tremors, and ataxia. It could also be important to more closely monitor the development and behavior of siblings of children who have fragile X syndrome, although such monitoring can be challenging, as more subtle symptoms of ADHD or social deficits can pale in comparison to the more severe symptoms of fragile X syndrome. Parents are at risk of overlooking these more subtle symptoms, and they could be unaware of any difficulties at home or at school. 3
Our study was limited by its retrospective design. In addition, all neurodevelopmental diagnoses were obtained from a review of the electronic medical record; patients were not directly evaluated for this study to confirm their neurodevelopmental diagnoses recorded in the medical record. Our database also represents a clinical population, so we would expect the rates of neurodevelopmental disorders to be higher than general population screening for involvement in the premutation or intermediate allele carriers. Specifically, we only reviewed cases in which fragile X testing was ordered by the attending pediatrician for clinical purposes. However, despite these limitations, given the wide range of neurodevelopmental disorders identified in our patients with intermediate and premutation range fragile X cytosine-guanine-guanine expansions, our data suggest that close monitoring of children with premutations may lend itself to important treatment options, such as early intervention programs for children with developmental delays and stimulant medication for ADHD. Further, although there have been no previously reported cases of tremor and ataxia in the pediatric age group, specific treatments for these problems, such as beta blockers, acetylcholinesterase inhibitors, and dopamine agonists, are also potentially available. 5,9 Further studies comparing pediatric patients with tremor and ataxia to older carriers with fragile X–associated tremor/ataxia syndrome are warranted.
The premutation and perhaps even intermediate repeat alleles may be important additive genetic factors to understanding the pathophysiology of complex neurodevelopmental disorders, including autism spectrum disorders and ADHD. Since our patients were derived from a clinical sample, unbiased identification of carriers of the premutation, perhaps through newborn screening, will clarify the frequency of developmental problems in carriers of both the premutation and also the intermediate alleles. In the future, identification of both premutation carriers at risk and their potential developmental or behavioral phenotypes could assist in early intervention and treatment and improved long-term outcomes. 31
Footnotes
Acknowledgments
This study was performed at the Mayo Clinic in Rochester, Minnesota, and it was presented as a platform session at the 2010 Pediatric Academic Societies Annual Meeting in Denver, Colorado.
Author Contributions
MMR made contributions to the conception and design of the study, acquisition of data, and analysis and interpretation of data; wrote the first draft of the article; and gave final approval to submitted versions of the article. RGV made contributions to the conception and design of the study, acquisition of data, and analysis and interpretation of data; mentored the first author on the first draft of the article and revised it critically for important intellectual content; and gave final approval to submitted versions of the article. DB-V made contributions to the conception and design of the study, acquisition of data, and analysis and interpretation of data; revised the article critically for important intellectual content; and gave final approval to submitted versions of the article. WEH made contributions to the acquisition of data and analysis and interpretation of data; revised the article critically for important intellectual content; and gave final approval to submitted versions of the article. SSV made contributions to the analysis and interpretation of data; revised the article critically for important intellectual content; and gave final approval to submitted versions of the article. CMS made contributions to the acquisition of data and analysis and interpretation of data; revised the article critically for important intellectual content; and gave final approval to submitted versions of the article. RJH made contributions to the conception and design of the study, acquisition of data, and analysis and interpretation of data; revised the article critically for important intellectual content; and gave final approval to submitted versions of the article.
Declaration of Conflicting Interests
Randi J. Hagerman, MD, is on the advisory council for fragile X treatment at Novartis. All other authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Randi J. Hagerman, MD, has received funding from Roche, Novartis, Seaside Therapeutics, Curemark, and Forest for treatment studies in fragile X syndrome or autism.
Ethical Approval
This study was approved by the Institutional Review Board of the Mayo Clinic.