
Abstract
Anti-N-methyl-
Anti-N-methyl-
At this point of time, the exact prevalence is not known; however, various centers have shown up to 4% among patients with encephalitis. 4,5 Dalmau et al 1 has reported 419 cases over 3 years and thus it seems to be a relatively frequent disorder. It may present as a paraneoplastic syndrome in ovarian and testicular tumors, neuroblastoma, and Hodgkin lymphoma. 1 Currently no pediatric prevalence data are available.
The most reassuring part of this disorder is its significant response to immunotherapy, 1 implying that early detection can significantly minimize associated morbidity and mortality.
This article describes the clinical presentation, diagnosis, and management of 11 children (up to 18 years of age) with anti-NMDA receptor encephalitis from a tertiary care teaching hospital in North India.
Method
The medical records of children diagnosed with anti-NMDA receptor encephalitis at a tertiary care teaching hospital in North India from January 2010 to December 2012 were retrospectively reviewed. Overall, 11 serologically proven cases presented between January 2010 and December 2012. Their clinical, diagnostic, and management features have been summarized.
The serological test used is a qualitative indirect immunofluorescence-based test. It is directed against antibodies to NMDA-type glutamate receptors with transfected cells acting as substrates. Titers 1:10 and above induce a fine granular cytoplasmic fluorescence with mild nuclear staining.
Results
Clinical Features
Age at presentation ranged from 2.5 to 18 years (mean: 9 years, median: 10 years). Both genders (1.2:1, female to male) were affected. The symptoms were insidious in onset with 80% (9 of 11) having a chronic or subacute presentation. The common modes of presentation were progressive extrapyramidal syndrome with global neuroregression in 45% (5 of 11), epileptiform encephalopathy in 27% (3 of 11), and an overlap between the 2 in 27% (3 of 11) (Figure 1). Preceding flulike illness within the last 1 month was present in 6 of 11 (55%) cases. Extrapyramidal features included orofacial and limb dyskinesia in 64% (7 of 11), generalized dystonia in 27% (3 of 11), hemidystonia in 9% (1 of 11), and mutism in 36% (4 of 11). Seizure semiology was varied, that is, generalized in 64% (7 of 11), myoclonic in 18% (2 of 11), atonic in 9% (1 of 11), and focal in 36% (4 of 11). Other associated clinical features included sleep disturbances like sleeplessness and excessive sleepiness in 18% (2 of 11); psychiatric manifestations like hallucinations and delusions in 9% (1 of 11); emotional lability in 18% (2 of 11); hyperactivity, aggression, or both in 18% (2 of 11); stereotypies in 9% (1 of 11) and autonomic instability in the form of hypotension in 9% (1 of 11). None of the patients had an underlying malignancy on a follow-up ranging from 6 to 18 months (Table 1).
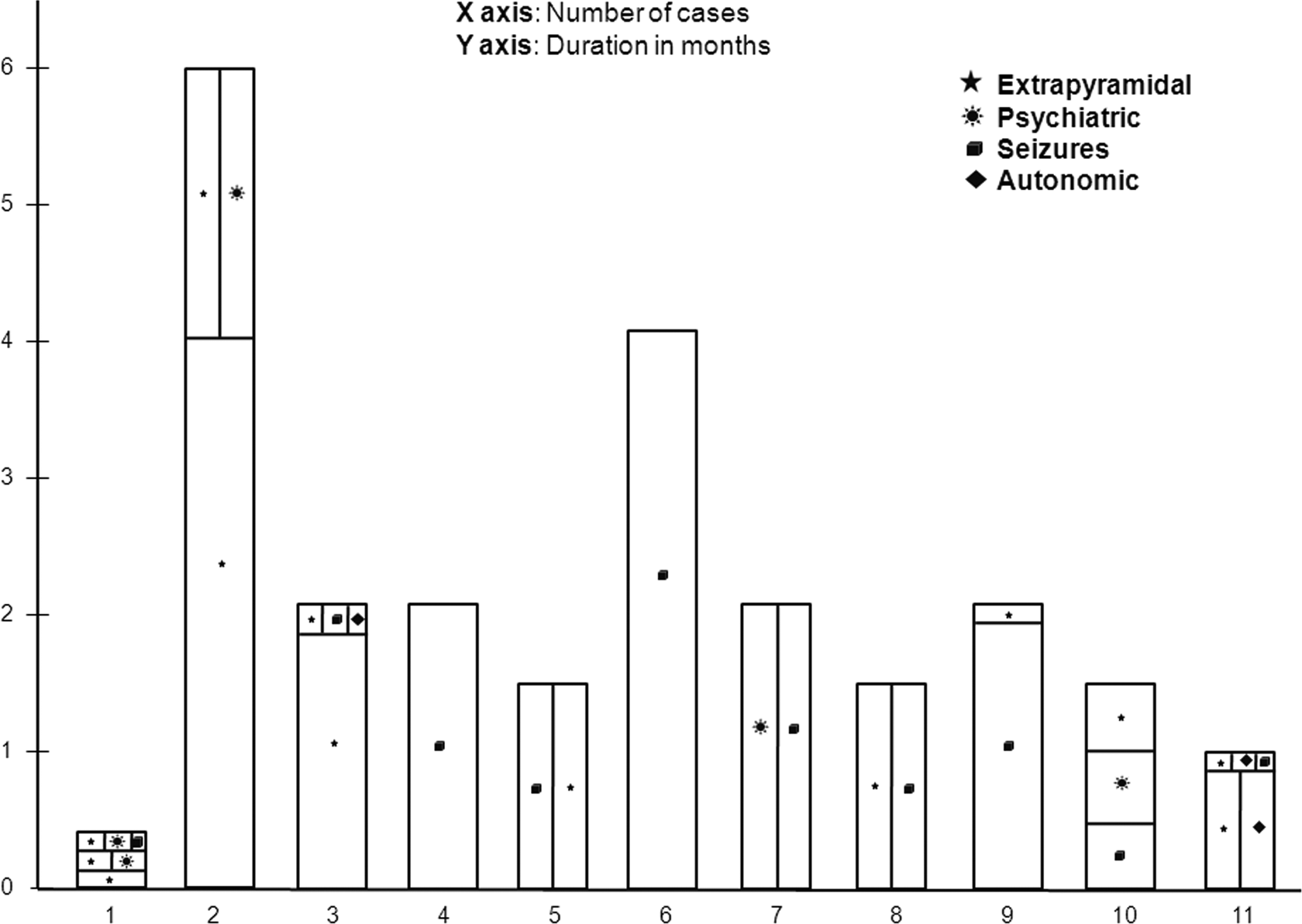
Graphical representation of evolution of symptoms in the cases before presenting at the current center.
Summary of Salient Clinical Features and Investigations in Cases.
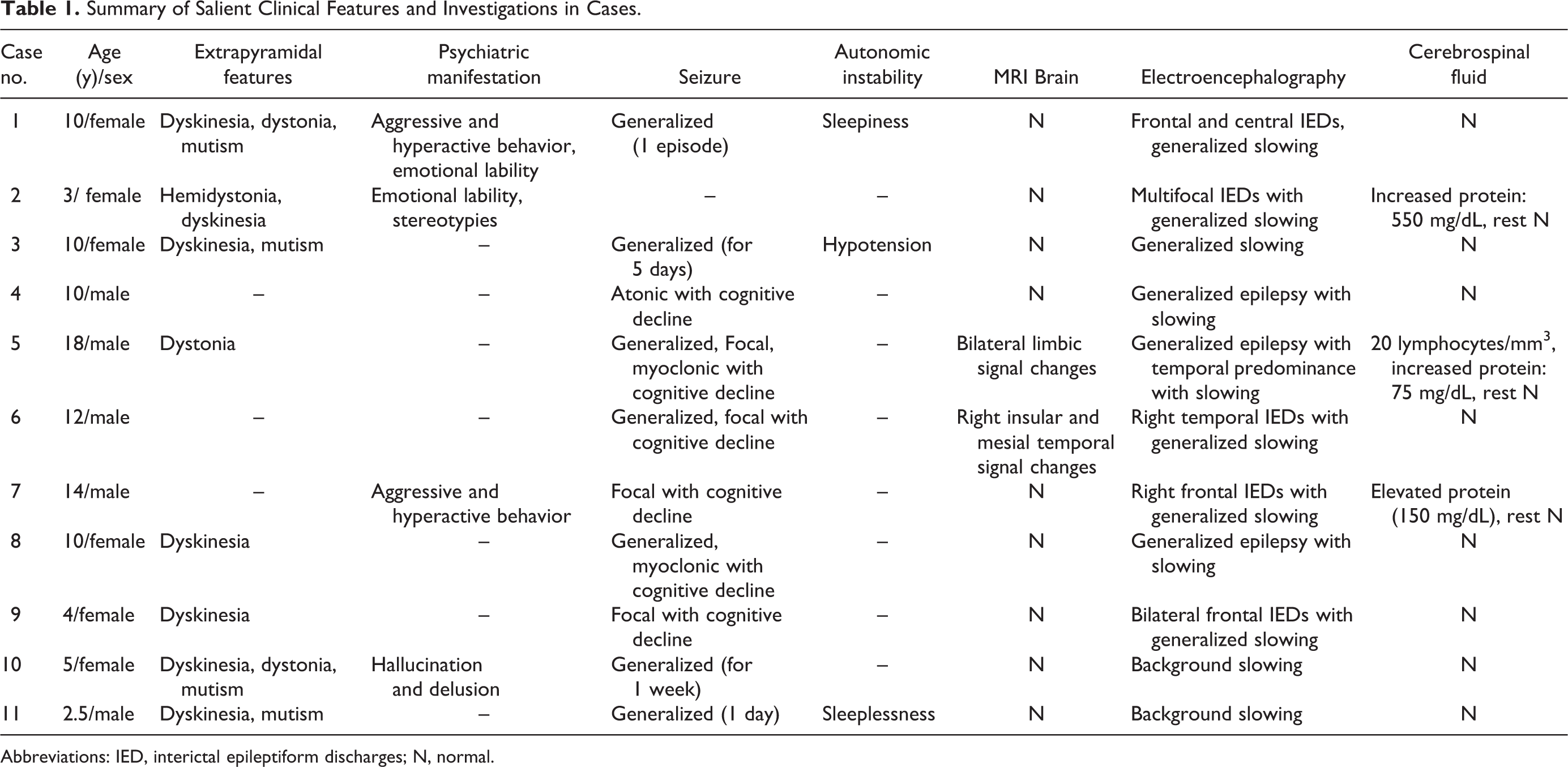
Abbreviations: IED, interictal epileptiform discharges; N, normal.
Investigations
All patients were positive for serum and cerebrospinal fluid anti-NMDA receptor antibody. Eight (73%) patients had normal cerebrospinal fluid microscopy and biochemistry, 3 patients had raised protein levels (75, 150, and 550 mg/dL), and 1 among them had 20 lymphocytes. Brain magnetic resonance imaging (MRI) was normal in all patients except 2 (18%), of which one had bilateral limbic signal changes and the other had right insular cortex and mesial temporal signal changes. Because of logistic reasons, follow-up brain MRI was not done in any of the cases to look at the radiologic evolution and remission.
Electroencephalographic (EEG) findings were nonspecific, which included generalized, multifocal, and focal (frontal and temporal) epileptiform activities in 8 (70%) patients with background slowing in all (Table 1).
Serum antinuclear antibody, anti–double-stranded DNA, anti–thyroid peroxidase antibodies, human immunodeficiency virus (HIV) status, metabolic workup (serum copper, serum ceruloplasmin, 24 hours urinary copper, arterial pH, blood glucose, arterial and cerebrospinal fluid lactate, blood ammonia, blood tandem mass spectrometry, urine gas chromatography–mass spectrophotometry), cerebrospinal fluid herpes simplex virus polymerase chain reaction), serology for Japanese B encephalitis, and peripheral smear for acanthocytes were done in relevant cases. None of the patients revealed any abnormality (Figure 2)
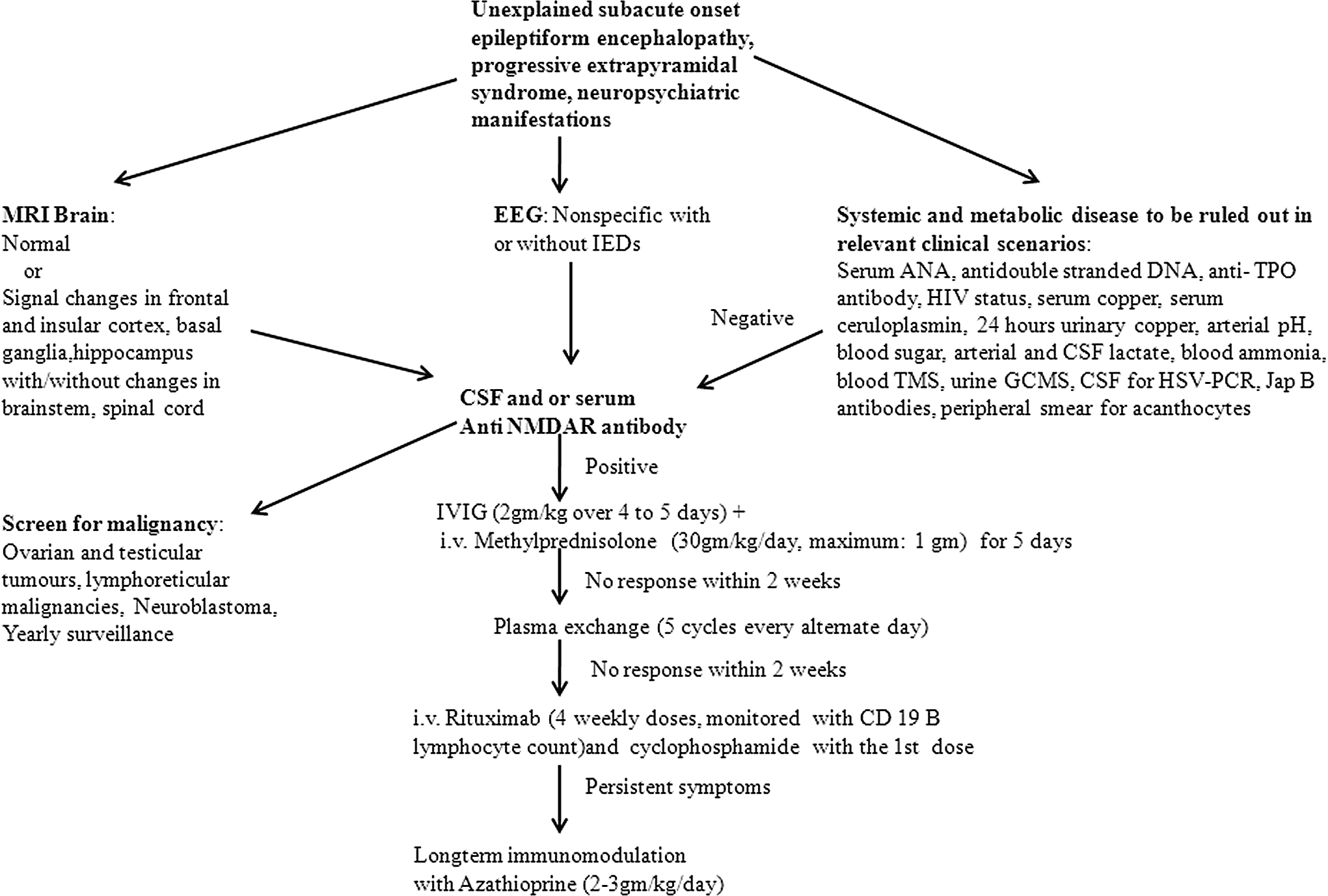
Flow chart depicting diagnostic and management approach in patients with suspected Anti-N-methyl-
Response to Treatment
All the patients received simultaneous steroid and intravenous immunoglobulin therapy. Five (46%) patients showed complete resolution to pulse methylprednisolone (30 mg/kg/d, maximum 1 g) and intravenous immunoglobulin (2 g/kg over 4 days) therapy followed by slow tapering of oral steroids (maximum 2 mg/kg/d, over 4-6 weeks) within a period ranging from 1 to 6 months. No relapse has been seen in any of these patients with maximum follow-up for 16 months. Two patients showed resolution of all symptoms except mutism; however, serial improvement has been documented. One patient presented with generalized and focal seizures with cognitive decline for last 4 months with evidence of right insular and mesial temporal signal changes on brain MRI. Even after right amygdalohippocampectomy and appropriate anticonvulsants, seizures persisted. In view of cognitive decline, anti-NMDA receptor antibodies were tested, which were positive. Only after immunotherapy the seizures responded, and currently the child has brief episodes of 1-2 focal seizures per month with significant improvement in cognition. Three patients (methylprednisolone and intravenous immunoglobulin failure) received plasma exchange (5 sessions every alternate day). Within 2 months, 2 patients showed complete resolution of symptoms. However, 1 patient who did not respond even to plasma exchange received 2 doses of weekly rituximab as well as 1 dose of cyclophosphamide without any improvement. Rituximab had to be discontinued in view of less than 1% CD19 B lymphocytes. In view of persistent anti-NMDA receptor antibody seropositivity, she received a second course of intravenous immunoglobulin, subsequent to which she had shown significant improvement (Table 2, Figure 2).
Summary of Response to Treatment of Cases.
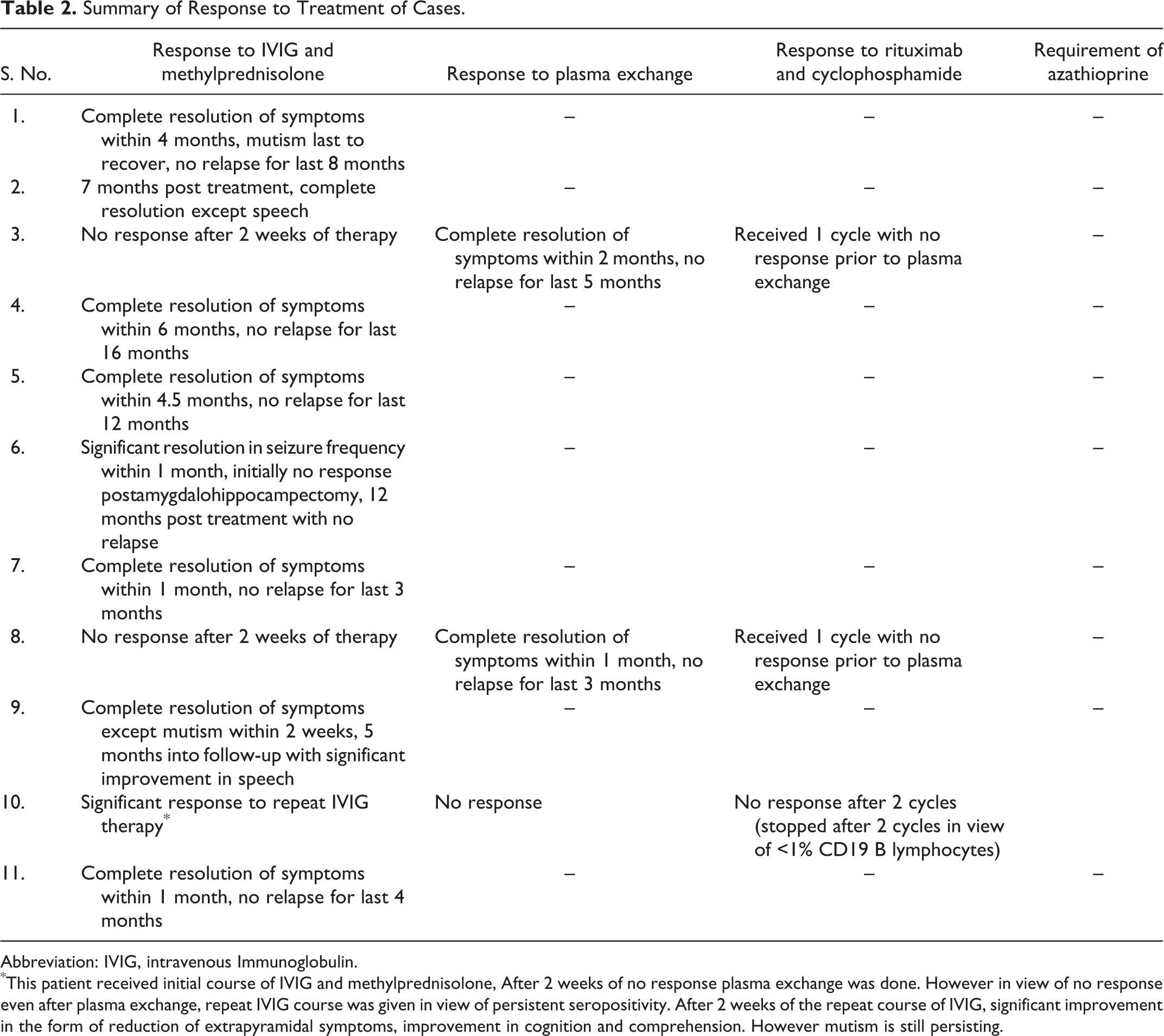
Abbreviation: IVIG, intravenous Immunoglobulin.
*This patient received initial course of IVIG and methylprednisolone, After 2 weeks of no response plasma exchange was done. However in view of no response even after plasma exchange, repeat IVIG course was given in view of persistent seropositivity. After 2 weeks of the repeat course of IVIG, significant improvement in the form of reduction of extrapyramidal symptoms, improvement in cognition and comprehension. However mutism is still persisting
Discussion
Anti-NMDA receptor encephalitis was first described in 2005 in 4 young women with ovarian teratomas who presented with neuropsychiatric and autonomic symptoms. 6 In the past 4 years, it has emerged as a significant entity post development of a radioimmunoassay by Dalmou et al. 1,7 Although primarily a disease of young adult women, it has been described in children. Overall, up to 60% cases have associated tumors, primarily ovarian teratomas, with pediatric patients (<18 years) having a tumor prevalence of less than 40%. 8 The current cohort reinforced this fact with female predominance and no evidence of malignancy in any of the patients till date.
In pediatric cases, the initial phase of neuropsychiatric manifestations can be nonspecific in the form of agitation, aggression, and temper tantrums. The first symptoms to be recognized in children are most often seizures, extrapyramidal features, and mutism. 1 These facts were evident in the current cohort. Interestingly, 1 patient had hemidystonia without any structural correlate. Unilateral movement disorder in association with anti-NMDA receptor encephalitis has been previously reported. 9 A subset of patients presented with subacute-onset epileptiform encephalopathy. The current recommendation states that autoimmune etiology should be ruled out in any subacute-onset unexplained epileptiform encephalopathy. 10 As compared to adults, autonomic symptoms are less common in children, as was seen in this group of patients. 1,8
The usual cerebrospinal fluid picture in these patients are lymphocytic pleocytosis, increased protein, and positive anti-NMDA receptor antibody. 11 Interestingly, in the current cohort, except for positive anti-NMDA receptor antibody in all, the cellular and biochemical response has been rarely noted. The antibodies could be absent in the serum post intravenous immunoglobulin or plasma exchange or in a protracted clinical course. Conversely, after recovery, antibodies could be detected only in the serum and not in cerebrospinal fluid. 4,12,13
The EEG findings are nonspecific. In the current cohort, uniformly all patients had slowing of background, a finding that is consistent with previous descriptions. 1,8 Three patients who did not have electrographic discharges had clinical seizures; however, seizures were a short-lasting phenomena in their disease process, which was primarily dominated by progressive extrapyramidal symptoms.
The brain MRI is abnormal in up to 50% of the cases. Signal changes are observed in the hippocampus, frontal and insular cortex, basal ganglia, brainstem, and occasionally spinal cord, which can or might not enhance on contrast administration. 7 This fact has been reinforced in 2 patients in the current cohort. In patients with recent-onset mesial temporal lobe epilepsy, workup for autoimmune encephalitis is recommended. 14 One of the reasons for low yield of MRI in the current group is that it was done early in the course of the disease and serial neuroimaging was not done.
In the current cohort, all except 2 patients responded to first-line immunotherapy (parenteral steroids and intravenous immunoglobulin). Verbal functions are one of the last to recover as has been reinforced in the current cohort. 1 Plasma exchange was done in the 2 nonresponders, with 1 showing complete resolution and the other showing no response. Pham et al 15 in their study indicated that earlier the institution of plasma exchange, better is the response, although this observation was trend based and the study was not adequately powered. However, it is an invasive procedure requiring expertise and intensive monitoring. 15 One patient who did not respond initially to first (including plasma exchange) and second line (rituximab and cyclophosphamide) of immunotherapy subsequently responded to a repeat course of intravenous immunoglobulin. Whether this is part of natural history of the disease or secondary to intravenous immunoglobulin is a matter worth debating. Although currently there are no recommendations, 1 it would be worthwhile to consider a repeat course of intravenous immunoglobulin, particularly in nonresponders, before starting relatively toxic long-term immunomodulators like azathioprine and mycophenolate.
As seen in this group, mortality is less than 5% with early institution of immunotherapy. 1 Spontaneous recoveries ranging from 6 months to 3 years have been reported; however, the recovery process is prolonged, with lasting neuromorbidities. 2 Relapses can occur in up to 25% of patients even more than a year after the initial episode. 11,12,16 None of the patients in the current cohort have suffered relapse, with the longest follow-up being 16 months.
Other autoantibodies implicated in central nervous system manifestations include anti–voltage-gated potassium channel antibodies like CASPR2 (contactin-associated protein 2) and LGI1 (leucine-rich glioma inactivated gene) and anti–glutamic acid decarboxylase antibodies. Among these, anti-LGI1 antibody encephalitis can have childhood presentation in the form of limbic encephalitis; however, it differs from anti-NMDA encephalitis by lack of extrapyramidal features (classically it is characterized by epileptiform encephalopathy and behavioral disturbances), signal changes in medial temporal lobes (in up to 70% cases), 8 and normal cerebrospinal fluid picture. 17 The only other encephalitis with movement disorder is an anti–voltage-gated potassium channel antibody encephalitis (faciobrachial dystonic seizures); however, it has an adult onset. 18 Among all autoimmune encephalopathies, anti–voltage-gated potassium channel has the best response to immunotherapy followed by anti-NMDA receptor with a partial and slow response and anti–glutamic acid decarboxylase with the worst response. 17 Other systemic autoimmune disorders associated with similar presentations include Hashimoto thyroiditis, lupus cerebritis, Sjögren syndrome, and antiphospholipid antibody syndrome. 19
The observations in the current case series cannot be generalized as the number of cases is not adequate for definite conclusions; however, they suggest a trend. Patient’s antibody status in serum and cerebrospinal fluid was not serially monitored; this could have depicted the clinicoimmunologic response to treatment. Moreover, the antibody assay was qualitative, so correlation with disease severity as well as various symptoms was not possible. All these features should be described in a large population of pediatric cases, so that definite conclusions can be drawn. Attempts should be made in future to unravel the specific predisposition at the level of human leukocyte antigen and major histocompatibility complex. Whether multiple autoantibodies can coexist in the same patient is a matter worth investigating.
In up to two-thirds patients with encephalitis like presentation, a specific infectious etiology is not found. 20 Autoimmune encephalitis should always be considered if common infectious etiologies are ruled out and there are specific clinical pointers in the form of unexplained epileptiform encephalopathy, neuropsychiatric manifestations, and a progressive extrapyramidal syndrome, particularly with no specific imaging or electrographic abnormalities. One should have a high index of suspicion as these disorders have favorable response to immunotherapy.
Footnotes
Author Contributions
All the cases were worked up under the supervision of SG and MT. BC prepared the manuscript and provided clinical care to the patients. SY and AKP provided clinical care to the patients and helped BC in preparing the manuscript. AS provided clinical care to the patients, particularly those requiring plasma exchange. BSR was involved in nursing care of the patients, including the plasma exchange patients. The final manuscript was approved by all the authors.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Ethical Approval
Ethical clearance was sought and approved by All India Institute of Medical Sciences, Institute Ethics Committee, New Delhi, India (approval number: IEC/NP-188/2013).