
Abstract
We present a case of a 16-year-old boy with Klinefelter syndrome who presented with a syndrome of impaired alertness, orofacial dyskinesias, choreiform movements, epileptic seizures, and autonomic instability, pointing to a diagnosis of anti-N-methyl-Daspartate (anti-NMDA) receptor antibody encephalitis.
Anti-N-methyl-
Case Report
A 16-year-old boy was referred to our hospital with a 6-day history of confusion and behavioral problems.
His medical history mentioned genetically confirmed Klinefelter syndrome. In addition, he was diagnosed with attention-deficit hyperactivity disorder (ADHD), for which he was treated with methylphenidate. The parents were nonconsanguineous. He had 2 sisters, one of whom had been treated for a benign ovarian teratoma.
Originally, he was admitted to another hospital, after developing paresthesias in the right foot and arm; he also developed speech problems described as a nonfluent language disorder with prominent word-finding problems. There were no other obvious clinical signs. Electroencephalography (EEG) showed a normal background rhythm and symmetrical increase of slow-wave activity. A computed tomographic (CT) scan of the brain was normal. Routine serum analysis and toxicologic screening ruled out metabolic abnormalities and intoxications. During his admission, the patient became aggressive, agitated, and restless, which prevented further testing of the patient. A nonorganic problem was suspected and the patient was transferred to our hospital about 1 week after symptom onset.
At admission in our hospital, he displayed a fluctuating alertness, with alternating episodes of somnolence and restlessness. He was well oriented in time and place, but because of his fluctuating alertness, he was at times unable to execute even simple verbal commands. His score on the Glasgow Coma Scale was 14. Routine neurologic examination was entirely normal, except for the presence of choreatic movements of the hands and face, which resulted in facial grimacing.
Laboratory testing, including serologic testing for antistreptolysin and antinuclear antibodies, did not show abnormalities. Toxicologic screening was once more negative. Repeat CT scan of the brain showed no cerebral lesions. A lumbar puncture was performed. Analysis of the cerebrospinal fluid was normal. Polymerase chain reaction results for varicella zoster virus, herpes simplex virus, cytomegalovirus, Epstein-Barr, and enterovirus were negative.
Because of uncontrollable agitation shortly after admission, we started treatment with olanzapine. Excessive agitation compromised follow-up clinical testing and the performance of neuroimaging.
Two days after admission, the patient became more somnolent and developed complex partial seizures with short-lasting episodes of generalized tonic posturing and head deviation. Treatment with valproic acid was started and the neuroleptic treatment was discontinued. Nevertheless, over the course of hours, he became unresponsive and was transferred to the intensive care unit. A control cerebrospinal fluid analysis now showed a cellular reaction (205 leukocytes/µL) and a slightly elevated protein content (65.2 mg/dL). Unexpectedly, sudden episodes of bradycardia and hypotension were seen during cardiac monitoring. Two times, asystole occurred, for which administration of atropine was needed.
Over the following days, the patient continued to have a fluctuating alertness (Glasgow Coma Scale score 3-8/15), perioral chorea, facial spasms, and choreiform movements of the right hand and foot. Meanwhile, repeated radiography of the chest had visualized a mediastinal mass (Figure 1A). A CT of the chest confirmed the presence of a probable mature teratoma (Figure 1B). The presence of anti-NMDA receptor antibodies in the patient’s cerebrospinal fluid confirmed the suspected diagnosis of autoimmune encephalitis (titer 1:3, cerebrospinal fluid 1:1).
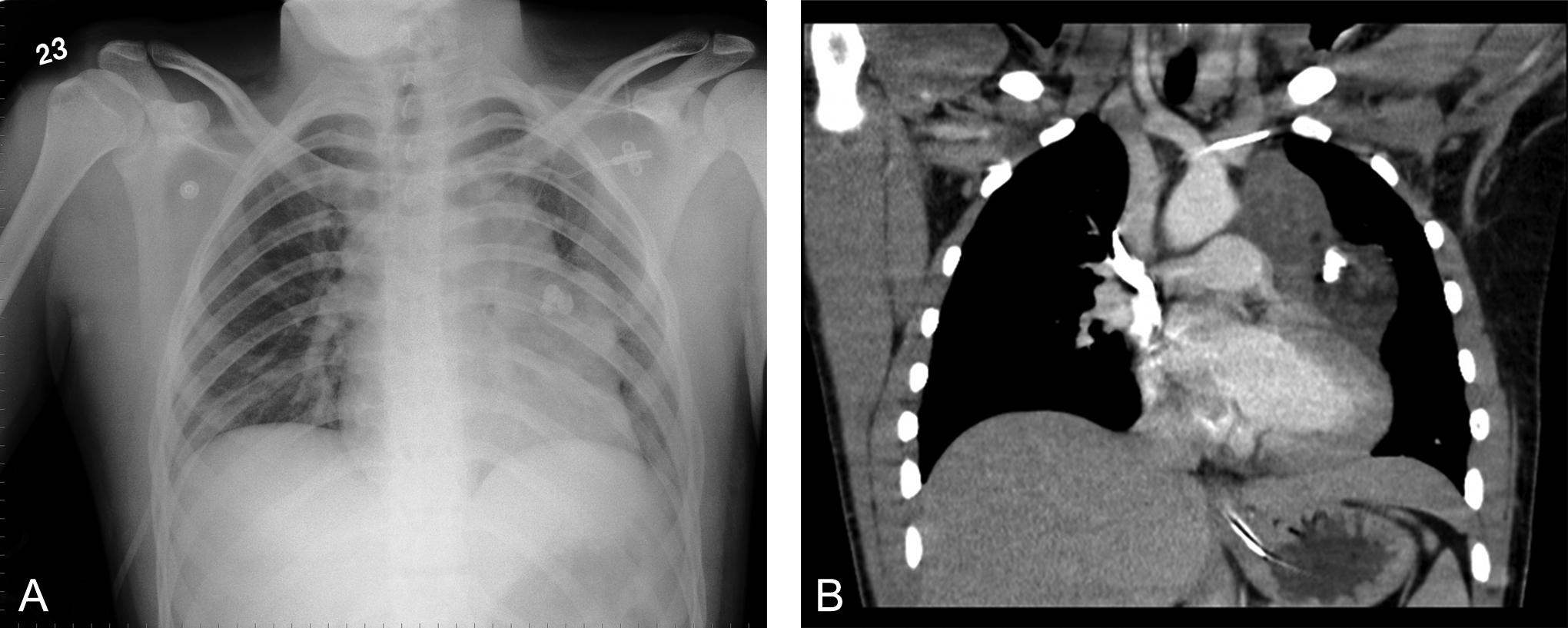
(A) Chest radiograph demonstrating a mediastinal mass. (B) Thoracic computed tomographic scan showing a mediastinal mass with calcifications.
A treatment with high-dose intravenous corticosteroids was started on day 6 after admission. Urgent mediastinoscopy and resection of the teratoma were performed 1 day later. Beta-human chorionic gonadotropin was elevated (5.04 IU/L), and alpha-fetoprotein was normal. Unfortunately, other endocrine investigations were not performed at that time. Anatomopathologic investigation confirmed the diagnosis of a mature teratoma. A teratocarcinoma was ruled out. In addition, 8 sessions of plasma exchange were done. As the condition of the patient was improving only slowly, cyclophosphamide pulse therapy was added (3 sessions of 500 mg with an interval of 1 month).
After 4 weeks of fluctuating alertness and infectious complications, the patient was transferred back from the intensive care unit to our department. At that moment, the patient opened his eyes spontaneously and was able to follow movements with the eyes. On the left side of his mouth and in both hands, myoclonic jerks were present. There was cogwheel rigidity in both upper limbs. He gradually improved and 1 month later, approximately, normal interaction with the patient was possible. He still had problems of dexterity because of postural and intentional tremor. After an admission of 4 months, he was finally discharged, dysplaying only mildly ataxic gait on clinical evaluation. Neuropsychological testing shortly before discharge were indicative of a relatively low verbal intelligence (IQ 74), reduced attention, and mild to severe verbal memory problems. Executive functions, for example, word fluency and alternating tasks, were below expected for his age. The problems of attention were supposed to be (at least partially) due to the preexistent diagnosis of ADHD. He had developed an excessive hyperphagia and rapidly gained weight and often felt tired. Serum titers of NMDA receptor antibodies at discharge were 1:200 (serum 1:20). He was still on oral steroids (32 mg), which were slowly tapered.
At reevaluation 6 months after onset, only a slight twitching of the fingers of the left hand was noted. The antibody titers in serum at that point was 1:200 (serum 1:20). Eleven months after his first presentation with us, we restarted immunosuppressive therapy (azathioprine 50 mg twice a day) as he developed disturbing disinhibited behavioral problems at school. As well, he had already gained 130 pounds because of hyperphagia. The neuropsychological testing was not repeated. The antibody titer in serum at that point was still 1:150 (serum 1:20). Every year, a whole-body PET scan is performed. Until now, a reoccurrence of tumor was not found, and the disinhibited behavior has improved. A dietician is helping him to lose weight.
Discussion
Anti-NMDA-receptor antibody–positive encephalitis is a rare neuroautoimmune disorder arising from the generation of antibodies targeting synaptic proteins. At first, it was considered a paraneoplastic disorder, most frequently associated with the presence of teratomas, as was also the case in this patient.
Most cases of anti-NMDA receptor antibody–positive encephalitis have been described in younger women with ovarian teratomas. Therefore, this case was interesting as it occurred in an adolescent with Klinefelter syndrome, which is known to be associated with a risk for teratoma. This boy had a rather normal pubertal development, which could, in retrospect, have been a clue to search for a teratoma in a patient with Klinefelter syndrome. 5
Peery et al 6 describe 4 different phases during an anti-NMDA receptor antibody–positive encephalitis: first, a prodromal phase (which was not present in our patient), followed by a psychotic and/or seizure phase, during which most patients, like ours, present at the hospital. After that, most patients develop an unresponsive phase and, finally, a hyperkinetic phase with autonomic instability (cardiac arrhythmia, hypotension, hypertension, hypoventilation, and hyper- or hypothermia). In children, as in our patient, the autonomic instability is less obvious than in adults, in whom it is one of the key signs. In our patient, we detected bradycardic episodes and asystole. In fact, the diagnosis has to be considered when a patient presents with a combination of psychiatric symptoms, autonomic dysfunction, and neurologic decompensation. Most often, before the final diagnosis is made, a number of differential diagnoses such as viral encephalitis, malignant neuroleptic syndrome, seizure disorders, or, as in our patient, psychogenic disorder is ruled out. Awareness is very important in this disorder, because other tests do not contribute to the diagnosis. Imaging of the brain (eg, magnetic resonance imaging) is aspecifically abnormal in 50% of the cases. Sometimes hyperintensities on T2 of fluid-attenuated inversion recovery images or contrast enhancement in nonspecific regions is seen. An EEG mostly shows nonspecific slow disorganized activity. The diagnosis rests on the detection of antibodies in the serum and the cerebrospinal fluid. 1,2,6
The classic treatment of this disorder consists of resection of the tumor, if present, followed by therapy with potent immunosuppressants, including high-dose intravenous corticosteroids, intravenous immunoglobuline, anti-inflammatory agents, plasma exchange, and monoclonal antibodies directed against CD-20 B-lymphocytes (eg, rituximab). These modes can be used in sequence or in combination. 7
The outcome of this syndrome is surprisingly good. After prompt adequate treatment, nearly 75% of the patients have complete recovery. 2 Executive functions and behavior are the most difficult to recover, as is the case in our patient. 8,9 Recurrences have been described in 20% to 25% of patients. 3 Benefits of early immunosuppression, tumor resection, and prolonged use of, for example, azathioprine have been described. 10
Footnotes
Acknowledgments
The patient was admitted to the University Hospital of Ghent, where we treated him. This case was presented at the VVN (Vereniging van Vlaamse Neurologen [Society of Flemish Neurologists]) autumn meeting in 2012 and a poster presentation was given at the World Congress of Neurology in September 2013.
Author Contributions
CS and PS were in charge of the patient during his admission. CS wrote the first draft of the manuscript and PS revised it for the final version.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Ethical Approval
Informed consent to publication was given by the patient and his mother (patient is below 18 years of age).