
Abstract
We present a 10-year-old boy with a predominantly motor multifocal neuropathy with demyelinating and axonal changes with sensory involvement, affecting only one upper extremity. Laboratory studies revealed an elevated titer of immunoglobulin M (IgM) antibodies against the NS6S antigen. He responded to treatment with high dose intravenous immunoglobulins. Focal or multifocal immune-mediated neuropathies are not common in children and may be underdiagnosed.
Keywords
Diagnosing focal neuropathic processes in children can be very challenging and recognizing a possible underlying autoimmune etiology is important as it may have major treatment implications. The spectrum of immune-mediated motor neuropathies is better characterized and recognized in adult patients but are less commonly diagnosed and rarely reported in the pediatric population. 1 –3
We present a case of unilateral, progressive upper extremity distal weakness in a child with predominantly motor multifocal neuropathy, with electrodiagnostic evidence of sensory involvement associated with NS6S disaccharide antibodies.
Case Presentation
A previously healthy 10-year-old right handed boy presented with a two-year history of insidiously progressive, painless, distal weakness in his left hand, predominantly affecting the ulnar innervated muscles. Despite severe weakness, progressive atrophy, and joint contractures, he never complained of any numbness or tingling. He denied any neck or shoulder pain. He denied any symptoms in his right arm or lower extremities. There was no history of any systemic disease and no history of trauma or infection preceding the onset of his symptoms.
His birth and developmental history were unremarkable. He did not take any medication and did not have any drug allergies. Family history was negative for similar problems or inflammatory diseases.
Prior to evaluation in our institution, he underwent a comprehensive assessment including extensive neuroimaging and multiple electrodiagnostic studies in other medical centers. Erythrocyte sedimentation rate, C-reactive protein, thyroid-stimulating hormone, Lyme titer, vitamin B12, and copper levels were within normal limits. Magnetic resonance images (MRIs) of the cervical spine, left brachial plexus and left forearm including were unremarkable. No significant abnormality to suggest entrapment of the ulnar nerve in the cubital tunnel or in the Guyon canal was observed.
He had prior nerve conduction studies and needle electromyograms (EMGs) in 2009, 2011, and 2012. They showed either unobtainable or severely reduced left ulnar compound muscle action potential and absent or severely reduced left ulnar sensory nerve action potential. Also the more recent studies showed slowing of median motor conduction velocity and near 50% conduction block in the median nerve in the forearm. Median and ulnar sensory and motor conduction studies from the right upper extremity were consistently unremarkable. EMG showed severe denervation of ulnar nerve–supplied muscles in the left hand, with no abnormalities in any muscle proximal to the wrist as well as neurogenic motor unit potentials in the abductor pollicis brevis muscle.
He was suspected of having a type 2 ulnar entrapment at Guyon canal with sparing of the dorsal ulnar sensory nerve. He underwent a surgical exploration and decompression at Guyon canal and the region around the wrist. However, his function did not improve and his weakness and muscle atrophy continued to progress; he was referred to our medical center for further evaluation.
On examination, no dysmorphic features were observed. His general medical examination was unremarkable. He did not have a rash or arthritis. Distal pulses were symmetric. On neurologic examination, he had normal mental status, speech, cranial nerves, cerebellar function, and gait. Motor examination showed striking atrophy of his left hand but retained grip strength graded 4/5 on the Medical Research Council (MRC) Scale. Strength testing of individual muscles of his left upper extremity: first dorsal interosseous 0/5, abductor digiti quinti 1/5, abductor pollicis brevis 5/5, and opponens pollicis 4/5. There was also possible mild weakness of the extensor indicis proprius (4 to 4+/5). Flexor contractures of the left fourth and fifth digits were observed. All other muscles in the left and right arm were 5/5. Sensory examination of the upper extremities was unremarkable. He had absent muscle stretch reflexes in the left upper extremity but normal (2+) reflexes in the right upper extremity. He had a normal motor and complete sensory examination of his lower extremities. Knee reflexes were relatively sluggish but the Achilles reflexes were 2+.
Another nerve conduction study/EMG was performed in our medical center. The nerve conductions study findings are summarized in Table 1.
Nerve Conduction Study Findings.a
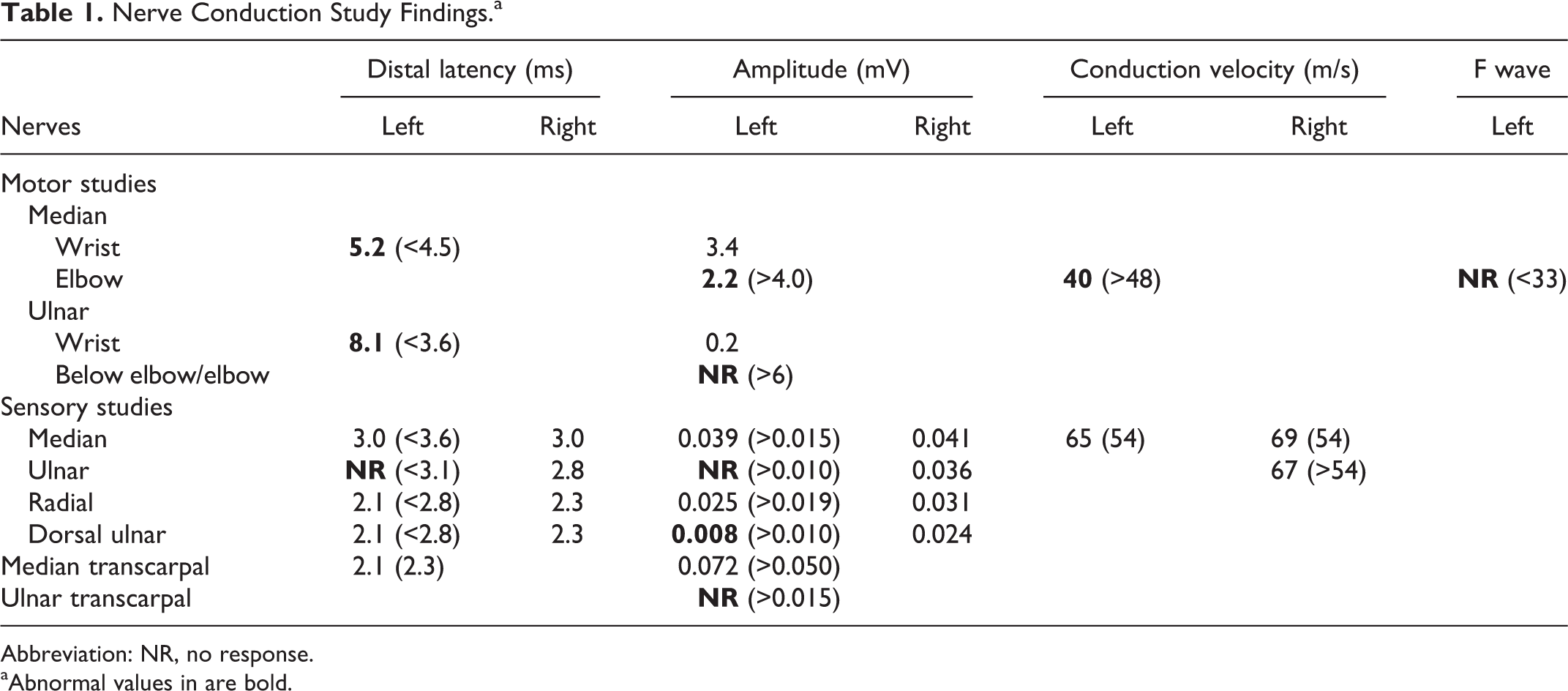
Abbreviation: NR, no response.
aAbnormal values in are bold.
On motor studies, there was severe reduction of the left ulnar compound muscle action potential, consistent with severe axonal loss. The prolongation of the distal latency, with such reduced, “residual” compound muscle action potential (0.2 mV) was likely caused by severe axonal loss but a demyelinating component was also possible. Similarly, absence of the ulnar compound muscle action potential with proximal stimulation is likely due to axonal loss and cannot indicate conduction block. The left ulnar sensory nerve action potential was absent. The dorsal ulnar cutaneous sensory nerve action potential was reduced compared to the right side, suggesting ulnar neuropathy proximal to the wrist, although specific localization was difficult. The median nerve compound muscle action potential was mildly reduced, with markedly prolonged latency and reproducible conduction block at the forearm segment. There was also some slowing of median conduction velocity. Median F waves were unobtainable. Despite severe abnormalities on the motor conduction studies, the sensory median nerve studies were completely normal, including comparison studies with the clinically unaffected contralateral upper extremity. Right ulnar and median motor nerve conduction studies were not repeated, as they were consistently normal in the past.
Needle EMG showed markedly diminished insertion activity, consistent with likely muscle fibrosis in the left hand ulnar nerve innervated muscles (first dorsal interosseous and abductor digiti quinti) and some spontaneous activity. No motor unit potentials were recorded from the first dorsal interosseous and only one motor unit potential was observed in the abductor digiti quinti with voluntary activation. Needle EMG in the left abductor pollicis brevis showed relatively mild active and chronic neurogenic changes. EMG of the ulnar innervated muscles proximal to the wrist and other muscles in the left upper extremity was completely normal.
The pattern of the electrodiagnostic changes indicated a multifocal, demyelinating and axonal process affecting the ulnar and median nerves on the left. The pattern of changes clearly indicated a postganglionic process making segmental anterior horn cell disease (monomelic amyotrophy) very unlikely. The findings, especially the diffuse demyelinating changes in the median nerve, including conduction block in the forearm, and normal EMG of lower plexus innervated muscles in the forearm made a lower brachial plexus lesion unlikely as well.
Given that the patient had a multifocal predominantly motor neuropathy, an autoimmune etiology was highly suspected. Serological testing for immune mediated neuropathy panel (Neuromuscular Laboratory Washington University, Saint Louis) showed that anti-GM1 ganglioside and myelin-associated antigen antibodies were negative. However the patient had moderately elevated IgM antibodies against NS6S disaccharide with a titer of 19 000 (moderately elevated range is considered between 15 000 and 30 000.)
Based on the multifocality of the neuropathic process and the significant demyelinating component, consistent with a likely immune-mediated process, the patient was empirically treated with a course of intravenous immunoglobulin at the dose of 0.5 g/kg/d for 4 days. On follow up exam examination three months later, there was evidence of improvement in some affected muscles. His left thumb abduction and opposition was 5/5, finger abduction was only minimally improved (1-2/5) likely because of advanced muscle atrophy and fibrosis, and fingers’ extension was 5/5. Repeat sensory examination revealed mildly decreased pinprick sensation over the fingertips of his fourth and fifth digits. He still had absent reflexes in his left arm. The rest of his neurologic examination was normal. Because of the patient stability on follow-up, the parents wanted to hold off any further immune therapy. Six months later, his left hand strength started to worsen again. A repeated nerve conduction study/EMG of the left upper and lower extremity was done at an outside facility. He had similar ulnar and median findings. There was no abnormality seen in the left lower extremity.
Discussion
Several neuropathic conditions are typically considered in the differential diagnosis of a child presenting with a unilateral progressive weakness and muscle atrophy in an upper extremity.
Early on, the presentation with weakness and atrophy in the distribution of a specific nerve can mimic an entrapment neuropathy syndrome. An ulnar neuropathy at the wrist was suspected and the patient underwent decompression of the ulnar nerve at the wrist without any improvement and continued to progress. Follow-up electrodiagnostic studies revealed a more diffuse, multifocal, axonal, and demyelinating process affecting both the ulnar and median nerves in the left upper extremity.
Painless Parsonage-Turner Syndrome/neuralgic amyotrophy, or brachial neuritis has been described in children. 4 However, the insidious gradual onset and the electrodiagnostic findings were not supportive of this diagnosis. Structural brachial plexus lesion or thoracic outlet syndrome were unlikely given his normal plexus MRI and the pattern of nerve conduction study/EMG findings. Possible monomelic amyotrophy, based on the pattern of distal weakness and atrophy, without sensory symptoms was a consideration, but the nerve conduction study/EMG was also not supportive of this diagnosis.
Inherited neuropathies such as hereditary neuropathy with liability to pressure were considered. Nonetheless, the localization of the median nerve conduction block in the forearm rather than a usual site of liability to pressure, the normal nerve conduction study/EMG in the right upper and left lower extremity as well as the lack of a family history make it unlikely so no further genetic testing was pursued.
Clinically our patient had a predominantly motor, multifocal neuropathy. The presence of significant, diffuse demyelinating changes in the median nerve, in addition to advanced axonal degeneration affecting the ulnar nerve, as observed on nerve conduction study/EMG strongly suggested an immune-mediated process. It was strongly suspected that he had either multifocal motor neuropathy with conduction block or multifocal acquired demyelinating sensory and motor neuropathy (Lewis-Sumner Syndrome).
Multifocal motor neuropathy with conduction block frequently affects distal regions of the upper extremity. It has been rarely reported in children. 1,2 Mild sensory symptoms or mild sensory deficits on examination are not unusual in patients with multifocal motor neuropathy with conduction block in the disease course. 5 Typically sensory nerve conduction velocities across the same segments with demonstrated motor conduction block have to be normal for the diagnosis of multifocal motor neuropathy with conduction block. 6 It is not clear how much sensory involvement is allowed for multifocal motor neuropathy with conduction block to be considered in individual cases. Hence, our patient does not fit the classic diagnostic criteria for multifocal motor neuropathy.
Multifocal acquired demyelinating sensory and motor neuropathy or Lewis-Sumner syndrome variant of chronic inflammatory demyelinating polyneuropathy is characterized by an asymmetric multifocal involvement and can start in the upper limb as initially described by Lewis et al. in 1982. 7 However, clinically, more sensory involvement would be expected and a monomelic form is not classically described.
Thomas et al. 8 described nine patients with focal upper limb predominantly demyelinating neuropathies. Reported patients had different combinations of motor and sensory nerve involvement and, in some patients, there were significant coexisting axonal changes. The authors believed that this syndrome represented a focal form of chronic inflammatory demyelinating polyneuropathy. Most patients had satisfactory response to immunotherapy, especially to intravenous immunoglobulin.
Although specific nosologic classification of our patient’s neurologic syndrome may be difficult, he appears to have a predominantly motor multifocal neuropathy, with mild sensory involvement in the spectrum of multifocal motor neuropathy with conduction block/multifocal acquired demyelinating sensory and motor neuropathy.
An immune-mediated etiology in patients with multifocal motor neuropathy with conduction block has been postulated based on the presence of different neuronal antigen antibodies in serum of reported patients. Serum IgM binding to GM1 ganglioside is associated with acquired motor neuropathies in contrast to chronic inflammatory demyelinating polyneuropathy, motor neuron disease, or sensory neuropathies. 9 However, in many patients with multifocal motor neuropathy with conduction block, anti-GM1 antibodies are not detected. 9,10 Anti-GM1 and anti–myelin-associated antigen antibodies were not detected in our patient. The elevated NS6S antibodies in our patient further supported an immune-mediated process.
The NS6S antigen is a peripheral nerve disaccharide. 9 Anti-NS6S disaccharide antibodies have been described in patients with motor neuropathies. Pestronk et al 9 reported that high-titer IgM antibodies to GM1 and NS6S each occurred in 43% of patients with motor neuropathies. Testing for IgM binding to NS6S in addition to GM1 increased the sensitivity for motor neuropathies from 43% to 64%. The authors did not identify any patients with amyotrophic lateral sclerosis or chronic inflammatory demyelinating polyneuropathy with high titers of IgM binding to GM1 or NS6S. High titers of NS6S antibodies were found less frequently in patients with sensory neuropathies and titers were lower compared to motor neuropathies. Patients with sensory neuropathies did not have high titers of IgM antibodies to GM1. Nobile-Orazio et al 10 reproduced these findings and found that testing for NS6S antibodies in addition to anti-GM1 IgM slightly increased the sensitivity of antibody testing in motor neuropathies (from 48% to 55%) but also reduced their specificity (from 95% to 85%) and positive predictive value for multifocal motor neuropathy (from 66% to 51%) because they were found in other autoimmune neuropathies. Our patient had moderately elevated antibody titers to NS6S that would be more characteristic of motor neuropathies versus chronic inflammatory demyelinating polyneuropathy, based on the aforementioned studies.
The discrepancy between the abnormalities observed on sensory nerve conduction studies and the lack of any significant sensory symptoms or signs in our patient is intriguing. It is possible that the immune process has been directed predominantly against the large fibers (motor and large, myelinated sensory fibers), not affecting the small sensory ones, and evolved very slowly, without causing sensory symptoms in the hand.
In conclusion, although immune-mediated neuropathies are not common in children, they may be underdiagnosed. Early recognition of potentially immune-mediated processes is essential in order to administer appropriate therapies as soon as possible to preserve the function and improve the neurologic deficit.
Footnotes
Acknowledgments
All workup and care was provided at the American Family Children’s Hospital, University of Wisconsin.
Author Contributions
FE and EN conceptualized and wrote the manuscript. FE provided clinical care to the patient. AW critically revised and reviewed the manuscript. AW also provided the detailed electro-diagnostic interpretation and reasoning.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.