
Abstract
X-linked Charcot-Marie-Tooth disease (CMTX) is the second common genetic variant of CMT. CMTX type 1 causes 90% of CMTX. The most important clinical features of CMTX are similar with other types of CMT; however, a few patients get the central nervous system involved with or without white matter lesions; males are more severely and earlier affected than females. In this review, the authors focus on the origin and classification of CMTX, the central nervous system manifestations of CMTX1, the possible mechanism by which GJB1 mutations cause CMT1X, and the emerging therapeutic strategies for CMTX. Moreover, several cases are presented to illustrate the central nervous system manifestations.
Keywords
X-linked Charcot-Marie-Tooth disease (CMTX) has a frequency of 7% to 15% among all CMT patients. 1,2 Like the classical CMT phenotype, CMTX is also characterized by slowly progressive distal muscle weakness and atrophy initially affecting the lower extremities (Figure 1), decreased or absent deep tendon reflexes, sensory abnormalities, and foot deformities (pes cavus and hammer toes), usually starting in the first 2 decades of life. However, unlike classical CMT, few CMTX patients have transient central nervous system symptoms and reversible cerebral white matter lesions.
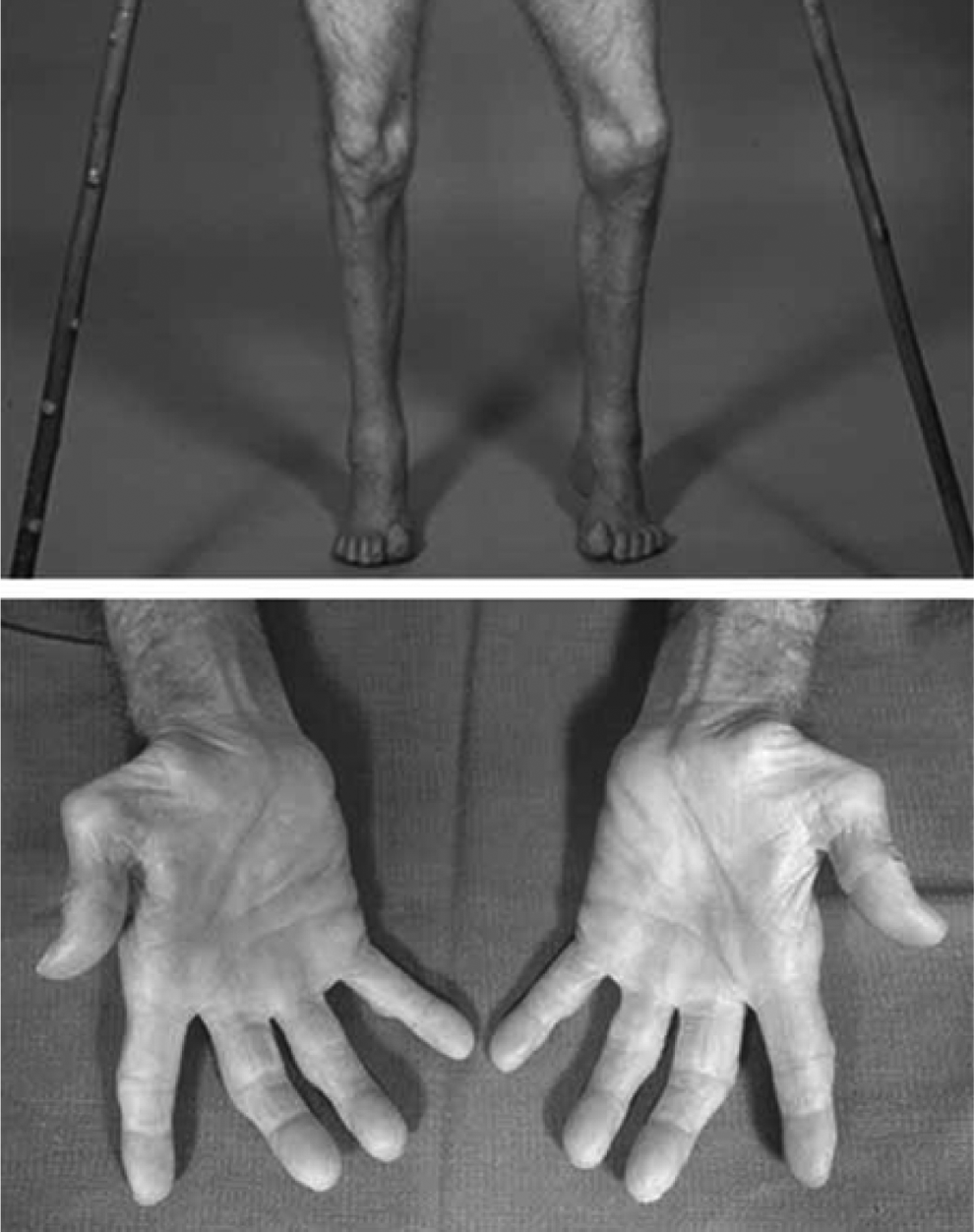
The patient was childhood-onset and examined at age 61 years with the Y211stop mutation on X-chromosome near Xq13. Note the advanced atrophy of distal muscles below the knee and in the hands with pes cavus. Source: patient IV.1, Hahn et al 1990. 12
CMTX can be classified into 6 types: CMTX1 (OMIM 302800), CMTX2 (OMIM 302801), CMTX3 (OMIM 302802), CMTX4 (Cowchock syndrome; OMIM 310490), CMTX5 (OMIM 311070), and CMTX6 (OMIM 300905). CMTX1 and CMTX6 are considered to have X-linked dominant inheritance, with male carriers being more strongly affected than females. Affected females usually have a later onset than males and a milder condition of the same phenotype at every age, or may even be asymptomatic, probably due to X inactivation in the myelinating Schwann cells. 3 –5 CMTX types 2-5 have X-linked recessive inheritance. CMTX1 may cause 90% of CMTX, and has become the second most common genetic variant of CMT after CMT1A. 6 In a recent clinical study of 245 patients in 116 CMT families, CMTX1 had a prevalence of 4.8% in all CMT patients, behind only CMT1A (19.6% prevalence). 7 Unlike CMTX1, all other CMTX forms are very rare.
CMTX1 is usually caused by mutations in the GJB1 gene on chromosome Xq13.1 encoding the gap junction beta 1 protein connexin 32 (Cx32), which has a length of 10 kb. 8 To date, more than 400 distinct mutations in the gap junction beta 1 gene have been identified. 9 Cx32 is a gap junction protein distributed in the peripheral nervous system and central nervous system as well as in the liver, kidneys, and pancreas. 10 GJB1 mutation has been reported to cause central nervous system lesions, as the gene is expressed not only in Schwann cells but also in oligodendrocytes. 11
In the following, the authors give a brief review of the origin and classification of CMTX, the central nervous system manifestations of CMTX1, the possible mechanism by which GJB1 mutations cause CMTX1, and emerging therapeutic strategies for CMTX. Moreover, the authors present several cases illustrating the central nervous system manifestations of the disease.
X-linked CMT
Origin of CMTX
In February 1886, Jean-Martin Charcot and his student, Pierre Marie, described 5 cases of progressive muscular atrophy in France. They assumed that the symptoms were caused by myelopathy, not neuropathy. In the same year, Tooth emphasized the early atrophy of the peroneus muscle and concluded that the condition was due to peripheral neuropathy in his thesis at Cambridge University. Shortly thereafter, in 1888, Herringham 13 identified a family in which males were selectively affected. Morgan’s demonstration of X-linked inheritance would not occur until 1910. In the next century, X-linked inherited neuropathy (CMTX) was described occasionally in sporadic kindreds. 14 –16 Its existence was momentarily questioned by Harding and Thomas in 1980. 17 To date, CMTX has now emerged as the second most common form of CMT after CMT1A, while CMTX1 accounts for 90%of CMTX cases.
Classification
So far, 6 types of CMTX have been found (Table 1). CMTX1 and CMTX6 have X-linked dominant patterns; the remaining 4 have X-linked recessive patterns. As has been noted, CMTX1 (OMIM 302800) accounts for 90% of CMTX, and has become the second most common genetic variant of CMT. CMTX2 (OMIM 302801) and CMTX3 (OMIM 302802) have been mapped to chromosomes Xp22.2 and Xq26.3-27.1, respectively. In 1 study, mental retardation was present in the family mapping to the CMTX2 locus, and spastic paraparesis was observed in families mapping to the CMTX3 locus. 18 CMTX4 (OMIM 310490), also known as Cowchock syndrome, has been mapped to chromosome Xq24-26.1 and is characterized by early childhood onset of a slowly progressive axonal sensorimotor neuropathy associated with sensorineural hearing loss and cognitive impairment. 19 CMTX5 (OMIM 311070) comprises the triad of optic atrophy, deafness, and polyneuropathy, and is caused by mutations in the phosphoribosyl pyrophosphate synthetase 1 gene (PRPS1) on chromosome Xq21.32-q24. CMTX6 (300905) is caused by a mutation in the pyruvate dehydrogenase kinase isoenzyme 3 gene on chromosome Xp22.11. Mild postural hand tremor has been recorded in CMTX6 patients. 2
Type, Subtype, and Gene Mutations of CMTX.
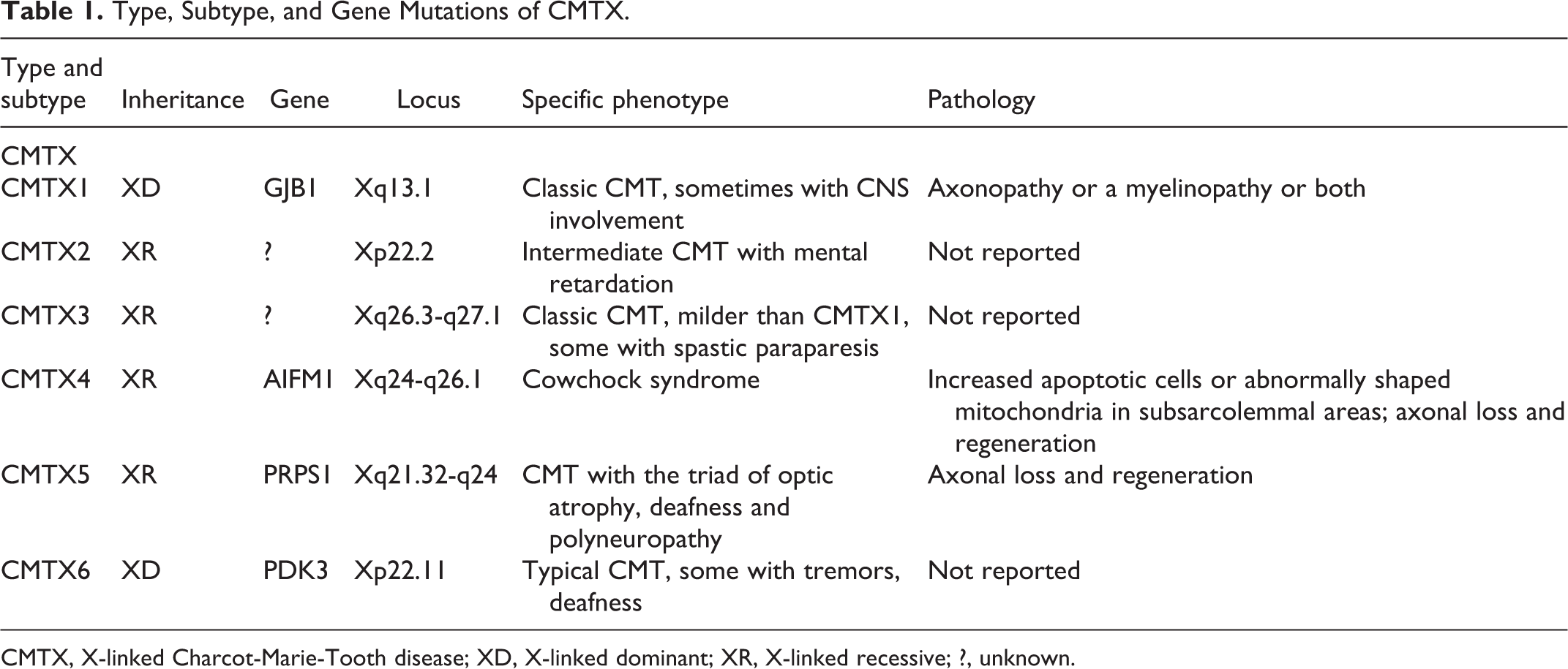
CMTX, X-linked Charcot-Marie-Tooth disease; XD, X-linked dominant; XR, X-linked recessive; ?, unknown.
GJB1 Mutation Causes CMTX1
The GJB1 gene is 1 member of the large connexin family encoding homologous proteins that form gap junctions in vertebrates. 20 GJB1 is responsible for encoding connexin 32, a 283-amino acid gap junction protein that is highly present in Schwann cells and oligodendrocytes, as well as a number of other cell types. Six connexins compose a hemichannel or connexon and arrange around a central pore. 20 Two hemichannels form a gap junction channel linking 2 cells. The channels allow the transport of ions, signaling molecules, and small metabolites and provide pathways for electrical and chemical coupling between the cells. 21,22 They also have a role in the regulation of cell growth and resistance to cell death. 23
Prior to the first report by Bergoffen et al 8 in 1993 showing 7 different GJB1 mutations in people from 8 CMTX families using direct sequencing, Gal et al 24 suggested that the gene responsible for CMTX1 is located on the proximal long arm of the X chromosome in 1985, and Kumar and Gilula 10 isolated the human CX32 gene from a human liver cDNA library in 1986. To date, more than 400 mutations in GJB1 have been described 9 and are thought to affect all regions of the connexin 32 protein. More than 300 mutations have been listed at http://www.molgen.ua.ac.be/CMTMutations/default.cfm.
It is still not well understood how GJB1 gene mutations lead to the characteristic features of CMTX1. Proposed mechanisms include loss of Cx32 function affecting the gap junctions in the myelin sheath and causing CMTX1 peripheral manifestations, 22,25 –27 or gain of function affecting the central nervous system. 22,23,26,28 Transgenic mice with the GJB1 gene mutation develop a loss of Cx32 function in Schwann cells and oligodendrocytes. 27 Nonsense or frame shift mutations that affect the N-terminus of Cx32 would not be expected to produce any functional channels. In addition, a few mutations likely abolish the expression of Cx32 by affecting the GJB1 promoter or the translation of Cx32 mRNA. 29 –31 Moreover, the entire coding region of GJB1 is deleted in several CMTX1 kindreds with central nervous system involvement. 25,32 Because different GJB1 mutations, including deletions, appear to cause a similar degree of neuropathy, most or all GJB1 mutations likely cause loss of function in myelinating Schwann cells, but the reverse in the central nervous system. Skewed X inactivation may explain why males are affected more severely and earlier than females, 3,4 although this has not been proven. The difference between CMTX1 and the other X-linked types was shown in Table 1, including clinical phenotype and pathology.
Central Nervous System Manifestations of CMTX1
In addition to the typical CMT clinical features, patients with CMTX can have delayed motor development, sensorineural hearing loss, tremors, pathologic fractures, recurrent central nervous system disturbance, or transient white matter lesions, which are reversible. 1 Generally, males tend to be affected earlier and more severely than females, with onset in the first 2 decades of life including walking difficulty, slowly progressive distal muscle weakness, and sensory abnormalities. In comparison, females show mild symptoms or are asymptomatic. Recent studies report that females carrying a GJB1 mutation have a variable phenotype: approximately two-thirds have a mild nonprogressive phenotype, one-third have a moderately severe phenotype that increases with age, and a small proportion are in a subclinical state. 3 Furthermore, 1 recent study suggested that the variable phenotype in females with CMTX1 may be partly due to the X inactivation pattern in myelinating Schwann cells. 6
After Nicholson and Corbett 33 reported abnormal prolonged brainstem auditory evoked responses in a few patients with CMTX1 in 1996, several CMTX cases with transient central nervous system involvement were reported and are now well recognized. In 2012, the authors 34 reported on a young man who presented with transient neurological manifestations, white matter lesions observed by cerebral magnetic resonance imaging (MRI) and atypical CMTX1 features associated with a new-found missense mutation (Asn54Ser) of the connexin 32 gene. As of this writing, 42 articles have been published on PubMed. 26,34 –39,41 –51,53,55 –61,63 –77 Among these cases, 74 patients had CMTX1 with central nervous system involvement and are presented in Table 2. In these cases, every central nervous system manifestation is a variant combination of dysarthria, dysphagia, hemiparesis, tetraparesis or complete paralysis, ataxia, cranial nerve deficits, expressive or motor aphasia, diplopia, vertigo, and dyspnea, and some have acute disseminated encephalomyelitis–like attacks. 35 Five main central nervous system phenotypes in CMTX patients have been reported: (1) subclinical abnormalities of visual-evoked responses and auditory-evoked responses, 33,53,55,62 (2) overt mild fixed abnormalities on neurological examination and/or central nervous system imaging that may or may not be accompanied by clinical manifestations, (3) severe transient central nervous system dysfunction accompanied by white matter changes observed by MRI, (4) mild to severe cognitive impairment, 43 and (5) persistent central nervous system manifestations. 47 Recently, Al-Mateen et al 41 conducted a study of 21 CMTX patients with central nervous system phenotypes. In his results, 17 patients had dysarthria, dysphagia, or both (81%); 13 patients had hemiparesis (62%); 9 patients had tetraparesis or complete paralysis (43%); and 7 patients had ataxia (33%). Other symptoms reported in less than 20% of the 21 cases included cranial nerve deficits, expressive aphasia, vertigo, and dyspnea. The first persistent central nervous system involvement case was described in 2009 by Siskind et al. 47 Central nervous system manifestations do not appear to correlate with the stage and severity of peripheral neuropathy; in some cases they are the initial manifestation of CMTX, while in others with exceptionally severe neuropathy, no clinical central nervous system phenotypes are present. The duration of central nervous system manifestation varies, but usually resolves between a few hours and a few weeks. However, cerebral recovery typically lags by months, as seen by MRI.
74 CMTX1 Patients With CNS Involvement.
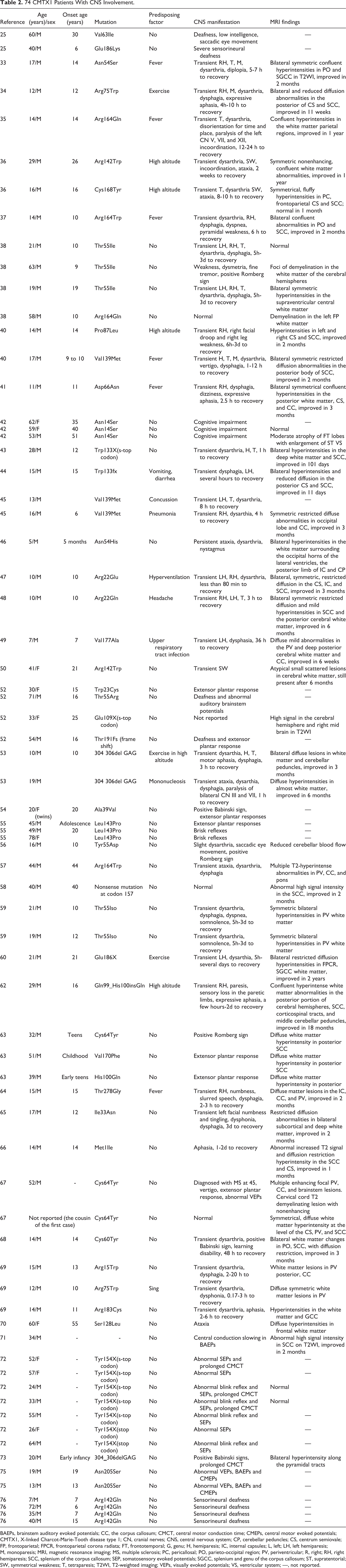
BAEPs, brainstem auditory evoked potentials; CC, the corpus callosum; CMCT, central motor conduction time; CMEPs, central motor evoked potentials; CMTX1, X-linked Charcot-Marie-Tooth disease type 1; CN, cranial nerves; CNS, central nervous system; CP, cerebellar peduncles; CS, centrum semiovale; FP, frontoparietal; FPCR, frontoparietal corona radiata; FT, frontotemporal; G, genu; H, hemiparesis; IC, internal capsules; L, left; LH, left hemiparesis; M, monoparesis; MRI, magnetic resonance imaging; MS, multiple sclerosis; PC, pericallosal; PO, parieto-occipital region; PV, periventricular; R, right; RH, right hemiparesis; SCC, splenium of the corpus callosum; SEP, somatosensory evoked potentials; SGCC, splenium and genu of the corpus callosum; ST, supratentorial; SW, symmetrical weakness; T, tetraparesis; T2WI, T2-weighted imaging; VEPs, visually evoked potentials; VS, ventricular system; —, not reported.
The pathological mechanism by which GJB1 mutations cause the central nervous system dysfunction in CMTX1 is not well understood, but may involve a decrease in the number of functioning gap junctions between oligodendrocytes and astrocytes, disrupting gap junction communication and leading to abnormalities in the ability of these cells to regulate intercellular fluid exchange of ions and small molecules. 9 Some authors have hypothesized that transient central nervous system manifestations in CMTX may be caused by reversible axonal damage in cerebral white matter, resulting from the expression of mutant connexin 32 in oligodendrocytes. 44 Aside from gene mutations, expression of the central nervous system phenotype in CMTX patients may be affected by other precipitating factors. In 1 study, 5 patients (24%) experienced symptoms after a fever, 4 patients (19%) after traveling to high altitude, 3 patients (14%) after exercise, 1 after a concussion, 1 after singing, and 1 after hyperventilation associated with emotional distress. 41
Based on white matter lesions seen in MRIs, various possible diagnoses include drugs, toxins, inherited metabolic disease such as adrenoleukodystrophy, or infectious and inflammatory causes such as acute disseminated encephalomyelitis and Guillain-Barré syndrome. Adrenoleukodystrophy is the most frequently inherited monogenic demyelinating disease. It can involve both the peripheral nervous system and the central nervous system. Initial MRI lesions usually involve the splenium of the corpus callosum and then extend into the adjacent white matter of the parieto-occipital and temporo-occipital lobes. Alternatively, initial lesions may involve the genu of the corpus callosum and then extend into the white matter of the frontal lobes. The cerebral demyelinating lesions are often symmetric, massive, and enhanced with contrast along the advancing edges. 78 The course of adrenoleukodystrophy is progressive, without complete recovery.
Acute disseminated encephalomyelitis usually occurs after a recent infection, and features an abrupt onset and a monophasic course. The lesions seen in cerebral white matter MRIs are usually asymmetric with spotty enhancement. Relapse or recurrent phases may occur 3 months after the first attack. 34 Guillain-Barré syndrome is also a monophasic disease, associated with either simultaneous or sequential cerebral demyelination. 79 Symptoms usually reach maximum severity after 4 weeks. 80
Transient paralysis may be seen in patients with periodic paralysis, alternating hemiplegia, Todd paralysis, moyamoya disease, mitochondrial encephalopathy with lactic acidosis and stroke-like episodes, and familial hemiplegic migraine.
CMTX1 should be included as a possible diagnosis for patients with transient central nervous system involvement and white matter lesions, as well as those who have peripheral neuropathy with reversible cerebral white matter degeneration.
A coexistent inflammatory demyelinating neuropathy has been described in patients with CMTX. 68,81 These observations indicate that overlapping inflammation in CMTX is probably not a rare phenomenon. The mechanism is unknown, but may include the exposure of myelin and/or Schwann cell antigens to which central tolerance has not been induced, which can then be presented to autoreactive T cells and trigger an autoimmune reaction against central nervous system myelin. 82
Therapy
Currently, there is no effective therapy available for CMT diseases, let alone specific treatment for CMTX. Some therapeutics have been used in an attempt to decrease the progression of CMT (especially CMT1 and CMT2), including ascorbic acid, progesterone antagonist, curcumin, 83 HDAC6 inhibitors, 84 and neurotrophic factors. 85 The most extensively studied therapy in CMT is ascorbic acid and progesterone antagonist. Ascorbic acid reduced the severity of neuropathy in transgenic mice overexpressing PMP22 (an animal model of human CMT1A) compared with untreated mice, 86 but several large trials with ascorbic acid in CMT1A patients were unsuccessful. 87 –91 It has been proven that progesterone antagonist can reduce PMP22 mRNA, slow the course of progressive muscle atrophy, and make neuropathological improvements in a transgenic rat model of CMT1A. 92,93 Unfortunately, current progesterone antagonists are too toxic to be safely administered to CMT1A patients, so developing a suitable analogue is necessary. Neurotrophin-3, a neurotrophic factor, was shown to be of possible minor benefit in a very small trial. 85 These findings need to be replicated in a larger trial. Curcumin 83 in CMT1B and HDAC6 inhibitors 84 in CMT2F have shown promising results in animal models, but have yet to be translated in humans.
On the other hand, as with other chronic neuromuscular disorders, there are many measures that greatly improve function and quality of life for CMT patients, such as physiotherapy, orthotics, and orthopedic surgery. Physical therapy and moderate activity to maintain as much strength and flexibility as possible are generally beneficial. 94 –96 Ankle-foot orthoses help control foot drop and ankle instability, improve physical functioning, reduce pain, and often provide a better sense of balance for patients. 97,98 Patients with limb and vertebral deformities often need corrective orthopedic surgeries. These interventions include soft tissue procedures, 99 arthrodesis, 100 –102 tendon lengthening or transfers, 103 –105 osteotomy, 106,107 and so on. An early minimally invasive approach includes plantar fasciotomy, tendon lengthening or transfer, transfer of the peroneus longus to the fifth metatarsal, and hammertoe repair of digits 1 to 5. 108 Wearing well-fitting footwear, trimming nails, removing calluses, keeping hands and feet warm, 109 not carrying extra weight, avoiding falls, using medication for pain, and obtaining psychological support and counseling can also improve the patient’s quality of life. Patients should also pursue careers that do not involve significant physical labor, as weakness is expected to worsen slowly over time.
In addition, neurotoxic agents such as vincristine, nucleoside analogs, cisplatin, carboplatin, and toxoids have been reported to worsen CMT 110 —mostly in CMT1A cases. One report described a girl with CMTX1 who was treated for Wilms tumor with chemotherapy, including vincristine. She tolerated vincristine with no evidence of neuropathy, raising the possibility that some patients with CMT may tolerate vincristine without severe adverse effects. 111 Another report presented a case of vincristine-related neuropathy that was exacerbated by voriconazole in a patient with previously undiagnosed, X-linked CMT. 112 While some CMTX1 patients may tolerate vincristine alone without neurologic side effects, there are insufficient data to comment on vincristine neurotoxicity in patients with CMTX1 and other less common CMT subtypes. In any case, neurotoxic agents should be avoided unless there are no reasonable alternatives, and until scholars gain a greater understanding of which CMT patients should not receive neurotoxic agents.
Genetic Diagnosis
Patients with CMTX usually have “intermediate” slowing of nerve conduction velocities, and mildly prolonged distal motor and F-wave latencies. 113 Nerve conduction velocities lie between CMT1 andCMT2. Together with a positive family history without male to male transmission, it should raise the consideration of CMTX in an appropriate clinical setting.
Most patients can obtain an exact genetic diagnosis. Genetic counseling can inform patients of the chance that they will pass CMTX on to their children. A female who is affected with an X-linked form of CMT has a 50% chance of passing down the condition in each pregnancy, no matter the sex of the child. A hallmark feature of X-linked inheritance is that fathers do not pass the condition on to their sons. However, since the father needs to pass on his X chromosome to have a daughter, all daughters born to a father with an X-linked form of CMT will be affected with the condition.
Conclusion
CMTX is the second most common genetic variant of CMT. The clinical manifestations of CMTX are varied and sometimes very atypical. Central nervous system manifestation may be the initial symptom of CMTX while other typical features are lacking. When the diagnosis is suspicious, careful clinical analysis and review of the literature may provide essential clues to the correct diagnosis. Furthermore, it is worth performing genetic testing and genetic counseling to arrive at a precise diagnosis and prognosis, and even more importantly to assess the risk of having children with CMTX. Finally, it is crucial that promising therapies are developed for this debilitating disease.
Footnotes
Author Contributions
YW drafted the initial manuscript, and reviewed and revised the manuscript. FY reviewed and revised the manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research and/or authorship of this article: This work was kindly supported by the National Natural Science Foundation of China (81370771) and the Hunan Province Key Technology Support Program (2015SF2019).
Ethical Approval
This study was approved by the Institutional Ethics Committee of Central South University.