
Abstract
Allan-Herndon-Dudley syndrome is a rare X-linked neurologic condition caused by mutations in monocarboxylate transporter 8 (MCT8), which leads to deficient thyroid hormone transport. Typical features include severe cognitive impairment, truncal hypotonia, spastic paraplegia, weakness, and speech difficulties. Minimal literature exists describing the ocular findings in patients with Allan-Herndon-Dudley syndrome. We describe 4 male siblings affected with Allan-Herndon-Dudley syndrome with a novel nonsense mutation (Q90X) in the MCT8 protein. All affected siblings presented with classic findings of Allan-Herndon-Dudley syndrome, and each of the siblings also developed intermittent esotropia. This group of affected siblings represents the first consistent documentation of strabismus in Allan-Herndon-Dudley syndrome, suggesting a possible association between this clinical finding and the neurologic syndrome.
Allan-Herndon-Dudley syndrome is an X-linked intellectual disability first described in 1944. 1 The syndrome develops as a result of a deficiency in monocarboxylate transporter 8 (MCT8), which has been mapped to the SLC16A2 gene. The MCT8 protein is implicated in the transport of triiodothyronine (T3) into cells, including neurons. Its deficiency leads to a hypothyroid state in the developing brain. 2 -4 The resulting and characteristic serum thyroid hormone levels include an elevated T3, a low or normal T4, normal or elevated thyroid-stimulating hormone, and a low reverse T3 (rT3). 2,4 The clinical picture of affected males begins at birth with hypotonia and muscle weakness followed by failure to meet developmental milestones. Profound intellectual disability is typical. The hypotonia progresses to spastic paraplegia and can include dystonic posturing of the hands. Other common neurologic complications include ataxia or failure to ambulate, seizures, and dysarthria or absent speech. 3,5,6 Although penetrance is complete, severity of complications varies depending on the mutation type. 2 The prognosis of patients with Allan-Herndon-Dudley syndrome also varies, with one case series of 32 patients reporting 8 patients who lived beyond age 70 and 3 dying before the age of 10. 3,7 Here we describe a family with a known and novel MCT8 mutation, with 4 of 5 male siblings affected with Allan-Herndon-Dudley syndrome. All 4 affected siblings developed intermittent esotropia, representing the first time strabismus has been consistently documented in association with Allan-Herndon-Dudley syndrome.
Case Summary
Four of the 5 male siblings born to a Hispanic mother were found to harbor a unique MCT8 mutation passed down from the carrier mother, causing Allan-Herndon-Dudley syndrome. The nonsense mutation was isolated to exon 1 of the SLC16A2 gene (c.268C>T, pQ90X). Two of the 4 affected males were half siblings (Figure 1).
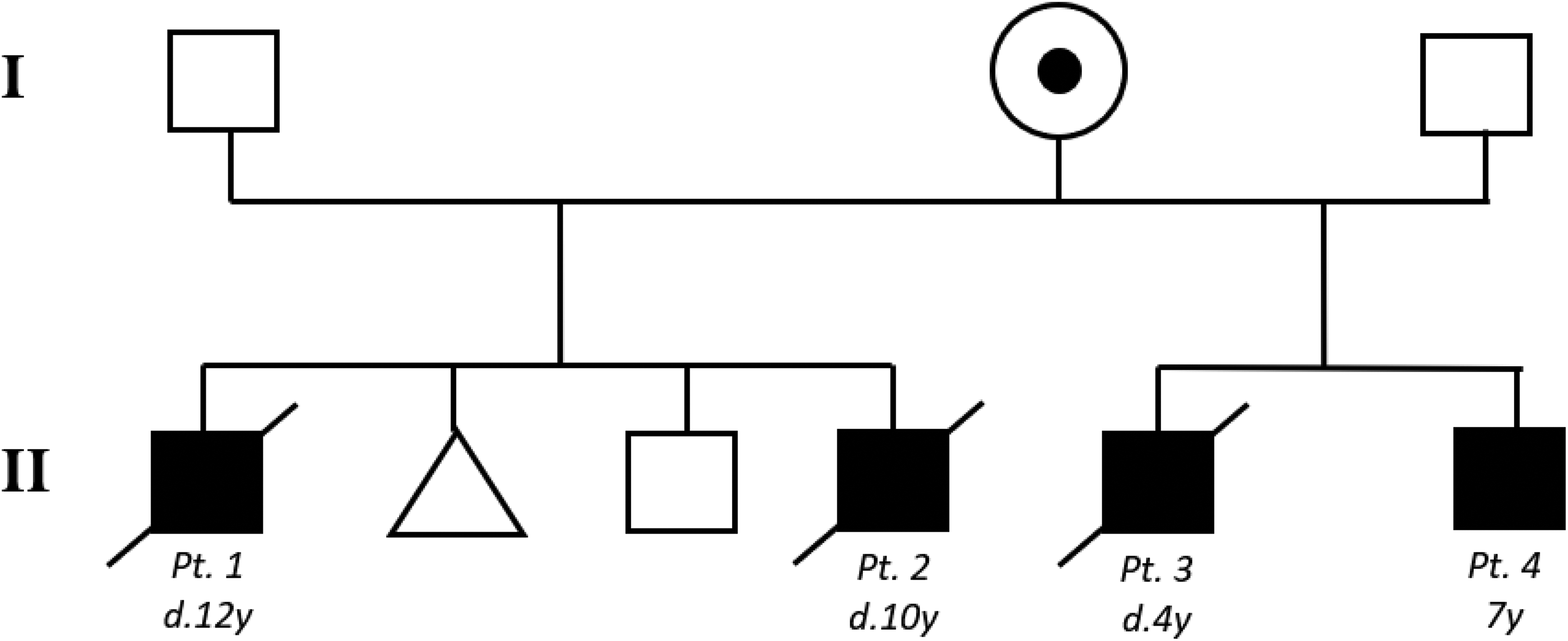
Kindred with Allan-Herndon-Dudley syndrome. Black squares indicate affected males. Dotted circle indicates the carrier mother. Triangle indicates spontaneous abortion. d, died; y, year.
Patients 1 and 2
The first sibship consists of 2 affected male siblings, born 4 years apart, a healthy male, and one spontaneous abortion. Patients 1 and 2 were born at 41 and 40 weeks, respectively, with uncomplicated pregnancies and had nearly identical Allan-Herndon-Dudley syndrome presentations. Each patient failed to meet gross motor developmental milestones with limited head control and an inability to roll, sit, or stand. Verbalizations beyond simple one-syllable words were never achieved, and each patient was wheelchair bound and nonambulatory. Both had severe cognitive impairment, microcephaly, as well as central hypotonia and peripheral hypertonicity. Seizures developed at approximately 2 years of age and were treated medically. Thyroid studies demonstrated a typical profile, including elevated T3, low T4, and normal thyroid-stimulating hormone. Magnetic resonance images were performed in both cases and demonstrated decreased white matter. Patients 1 and 2 died at ages 12 and 10 from complications of Allan-Herndon-Dudley syndrome, respectively.
Patients 3 and 4
The second sibship consists of 2 affected male siblings. The first (patient 3) presented with severe cognitive impairment and failure to progress developmentally. He also was born with central hypotonicity and peripheral hypertonicity and died by age 4.
Patient 4 had extensive evaluation at our facility starting at age 6. He was born premature at 36 and 3/7 weeks’ gestation and required a 2-week hospitalization for pulmonary maturation and neonatal jaundice. The pregnancy was complicated by maternal gestational diabetes. The patient had global developmental delay and was never able to sit without support. His body mass index has been in the 8th percentile. He is nonambulatory and wheelchair bound. Vocalizations beyond simple 1- to 2-syllable words have not developed. The patient’s other neurologic symptoms are consistent with the Allan-Herndon-Dudley syndrome and include spastic quadriplegia, truncal hypotonia, extensor posturing, generalized myoclonic epilepsy, and severe cognitive impairment. The patient’s thyroid hormone panel was also typical of the Allan-Herndon-Dudley syndrome phenotype.
The patient had a full ophthalmologic evaluation by a pediatric ophthalmologist in response to a failed vision screen and reports of intermittent inward turning of the eyes. The patient had fix and follow vision in each eye. He had an intermittent right esotropia of 10-12 prism diopters by prism alternate cover test with latent nystagmus in each eye. Cycloplegic refraction revealed moderate hyperopia of +2.00 diopters with mild astigmatism of +0.75 diopters in both eyes. Posterior examination showed pink, normal-sized, flat optic discs with cup-to-disc ratios of 0.5 in both eyes. The anterior segment examination was normal. The foveas, vasculature, and peripheral retina all appeared normal. The motility was full. The patient’s esotropia did not improve with glasses. The parents noted that the other 3 affected siblings wore glasses and developed intermittent turning in of the eyes. Glasses did not improve the esotropia in the siblings and none had strabismus surgery. There was no family history of strabismus in either of the parents or extended family.
Discussion
We present a case of a family with a novel MCT8 mutation (c.268C>T, pQ90X) in a carrier mother and 4 male siblings affected with Allan-Herndon-Dudley syndrome. All 4 affected siblings developed classic neurologic features of Allan-Herndon-Dudley syndrome, as well as intermittent esotropia, which was not found in the unaffected male sibling. There was no improvement in strabismus with glasses in our patient or in the siblings by history, indicating a nonaccommodative strabismus. Given the intermittent nature and small angle of esotropia, strabismus surgery was not performed. Surgery was also not indicated given the risk of overcorrection with underlying neurologic disease
Literature describing eye findings associated with Allan-Herndon-Dudley syndrome is lacking. Heuer reported in 2007 that strabismus and nystagmus are common findings in patients with MCT8 mutations. 8 However, only 2 cases of strabismus are reported. Both cases also had differing presentations from our patient. Wemeau et al discussed a case of divergent strabismus in a 16-year-old male patient with a missense mutation in exon 3 of the MCT8 gene, and Azzolini et al reported bilateral convergent strabismus in an Allan-Herndon-Dudley syndrome patient with a frameshift MCT8 mutation causing a premature stop codon and a truncated protein. 9,10 It may be that the presence of strabismus and, in this case, intermittent esotropia, is related to the degree of MCT8 transporter deficiency, with more severe genotypes such as those in our series with truncated and nonfunctional proteins causing more significant neuronal pathology and more severe hypertonicity. This hypothesis is supported by the prognosis in these siblings, as none of the affected individuals have survived into their teenage years, suggesting a severely pathologic phenotype. Our case strengthens the association between Allan-Herndon-Dudley syndrome and intermittent esotropia, linking the probability of its development to genotypic severity.
Footnotes
Author Contributions
DN served as the pediatric ophthalmologist who cared for patient 4; he also edited the manuscript. CS prepared the first draft of the manuscript and edited the article thereafter.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Ethical Approval
Approval for publication was provided by the guardian of the patient described in this case report. IRB approval at our institution was not required given the nature of the case report.