
Abstract
Objective:
We performed a retrospective chart review of patients with trisomy 21 and infantile spasms in our university-based pediatric epilepsy center between 2002 and 2016 in order to describe the clinical characteristics of children with these diagnoses as well as to evaluate their response to first-line treatments.
Methods:
Patients with infantile spasms were identified via the neurophysiology database. Charts were reviewed with attention to infantile spasms diagnosis, presence of trisomy 21, age of reported clinical onset, treatment lag, treatments used, response to treatment, imaging findings, electroencephalography (EEG) data, and developmental outcomes.
Results:
Of the 310 patients with infantile spasms, 24 also had trisomy 21. Three patients did not meet inclusion criteria. Ten of the 21 patients received nonstandard therapies first line; 2 of the 10 (20%) achieved spasm control, and 4 of the 8 who failed therapy (50%) progressed to Lennox-Gastaut syndrome. Eleven of the 21 patients received standard therapies as first-line treatments (10 with prednisolone according to the protocol in the United Kingdom Infantile Spasms Study [UKISS] and 1 with adrenocorticotrophic hormone [ACTH]). Nine of the 10 patients (90%) who received prednisolone achieved spasm resolution, 6 (60%) of these without relapse. The final patient (10%) failed prednisolone as well as ACTH. One patient received ACTH first line with success.
Conclusion:
This is the only series to follow children with trisomy 21 and infantile spasms in which a significant proportion received UKISS-protocol prednisolone. It adds to current knowledge about safety, tolerability, and effectiveness of prednisolone in this group.
Epilepsy occurs in 5% to 13% 1 –3 of children with trisomy 21, and infantile spasms is the single most common epilepsy type in these infants, occurring in 6% to 32%. 3,4 The International League Against Epilepsy classifies infantile spasms into genetic, metabolic, infectious, immune, structural, or unknown subtypes 5,6 ; infantile spasms in children with trisomy 21 is classified as genetic in etiology. Vigabatrin or hormonal therapies such as adrenocorticotrophic hormone (ACTH) and prednisolone according to the protocol in the United Kingdom Infantile Spasms Study (UKISS) are considered first-line therapies for infantile spasms. 7,8 Since the publication of the UKISS experience in 2004, the use of high-dose prednisolone has increased and approached the use of ACTH as a first-line agent. 8 –10 Although some studies have noted the possibility that children with trisomy 21 respond best to ACTH, 9,10 controlled trials have not been carried out. Some children are given valproic acid first-line, their clinicians often citing a retrospective case series in which 9 patients had trisomy 21 and infantile spasms, 3 5 of whom (56%) were reported to respond to valproic acid, but the study does not define response to therapy, and age at last follow-up is not reported. There is also no report about recurrence with this therapy.
In this retrospective case series, we report the outcomes of standard first-line therapies for infantile spasms compared with other treatments in children with trisomy 21. Although this study’s primary outcome measure is clinical seizure remission, we are particularly interested in describing the adverse effects in this population as well.
Methods
This study was performed after obtaining approval from the University of Texas Southwestern Medical Center’s institutional review board. A retrospective chart review was performed at Children’s Medical Center, the tertiary care pediatric hospital of the University of Texas Southwestern Medical Center in Dallas, Texas. Patients were identified by querying our Excel-based neurophysiology database between 2002 and 2016 with keywords hypsarrhythmia, electrodecremental segment, spasms, West, and infantile. This was applied to inpatient and outpatient electroencephalograph (EEG) reports and prolonged video-EEG monitoring studies.
Inclusion criteria included confirmation of diagnoses of trisomy 21 and infantile spasms and at least 1 follow-up data point at our institution in an outpatient clinic. Exclusion criteria were lack of follow-up at our institution or severe structural abnormality to account for etiology of infantile spasms.
Clinical records were reviewed by a single reviewer, including the molecular diagnosis of trisomy 21, initial therapy for infantile spasms, treatment lag, response to therapy, reports of the first EEG diagnostic of infantile spasms as well as follow-up EEG, baseline magnetic resonance imaging (MRI), possible progression to Lennox-Gastaut syndrome (LGS), and developmental outcome. Treatment lag was defined as the interval between the reported onset of clinical spasms and the first treatment for infantile spasms.
Treatment responders were defined as patients with resolution of clinical spasms at 2-4 weeks posttreatment, many of whom had follow-up EEGs documenting resolution of hypsarrhythmia, though the timing of these follow-up EEGs varied by physician. Of the patients who responded to therapy, those who experienced subsequent relapses either during taper of hormonal therapy or weeks to months later are classified as responders with recurrence. The EEG reports were reviewed. The term hypsarrhythmia was used when the following criteria were met: high-voltage slowing, disorganized background, and multifocal sharp waves. 11 The term modified hypsarrhythmia was used according to the Hrachovy criteria. 12
Lennox-Gastaut syndrome was defined as the presence of multiple seizure types, including tonic seizures, and the presence of an EEG with slow spike and wave discharges, with or without electrodecremental segments or generalized paroxysmal fast activity.
There are no validated developmental scales specifically for children with trisomy 21 13 –15 ; therefore, we applied a developmental scale not specific to trisomy 21 and based on developmental quotient, retrospectively. In patients whose documented current skills at the last follow-up visit were expected for an age less than half their chronological age (a developmental quotient of approximately <0.40), the term severe delay was applied. For those functioning at a developmental level that would be appropriate for a child at half their chronological age (a developmental quotient approximately 0.4-0.75), the term moderate delay was assessed. For those with some delay, which did not approach the severe or moderate benchmark (a developmental quotient approximately >0.75), the term mild delay was given. In those patients with mention of delay but without further details as to current skill level, the term unspecified delay was given. A pediatric neurologist or pediatric geneticist documented the follow-up developmental information analyzed. The elements documented were frequently in qualitative form, rather than being documented with a standardized developmental scale. When there were discordant domains, the most delayed development was scored.
Nonhormonal antiseizure medications (ASMs) were dosed at the discretion of the treating neurologist. All patients who received prednisolone received either 10 mg 4 times daily (UKISS dosing) 7 or 15 mg 3 times daily (modified UKISS dosing). ACTH was given at a dose of 150 units/m2/d intramuscularly for 2 weeks and then weaned over the following 6-8 weeks. Urine glucose, stool occult blood, and blood pressures were monitored during treatments with hormonal therapies.
Nonparametric comparisons were analyzed with Fisher exact test for 2 independent groups using Stata, version 14 (Stata Corp LP, College Station, TX). A 2-sided P value of less than .05 was considered statistically significant.
Results
Clinical Presentation
Among the 480 patients who were identified through our neurophysiology database queries, 310 patients were confirmed to have infantile spasms. We identified 24 patients with infantile spasms and trisomy 21, accounting for 7.7% of the cases of infantile spasms at our institution in the 14-year review period. All children with trisomy 21 had molecular diagnosis confirming nondisjunction, with a single patient found to have mosaic trisomy 21.
One of the 24 children suffered severe neonatal meningitis with subsequent development of infantile spasms during the same admission; he ultimately died of complications of his infection prior to hospital discharge. He was treated with topiramate and did not receive hormonal therapies. He is not included in our analysis. Two children had EEGs performed as part of an acute inpatient hospitalization, but had clinical follow-up at outside institutions and did not meet inclusion follow-up criteria. They are excluded from our analysis. This left 21 patients in our series analyzed below and summarized in Table 1. The median age of onset was 7 months with an interquartile range of 4.5-9.5 months.
Clinical History, Diagnostic Workup, and Response to Treatment.
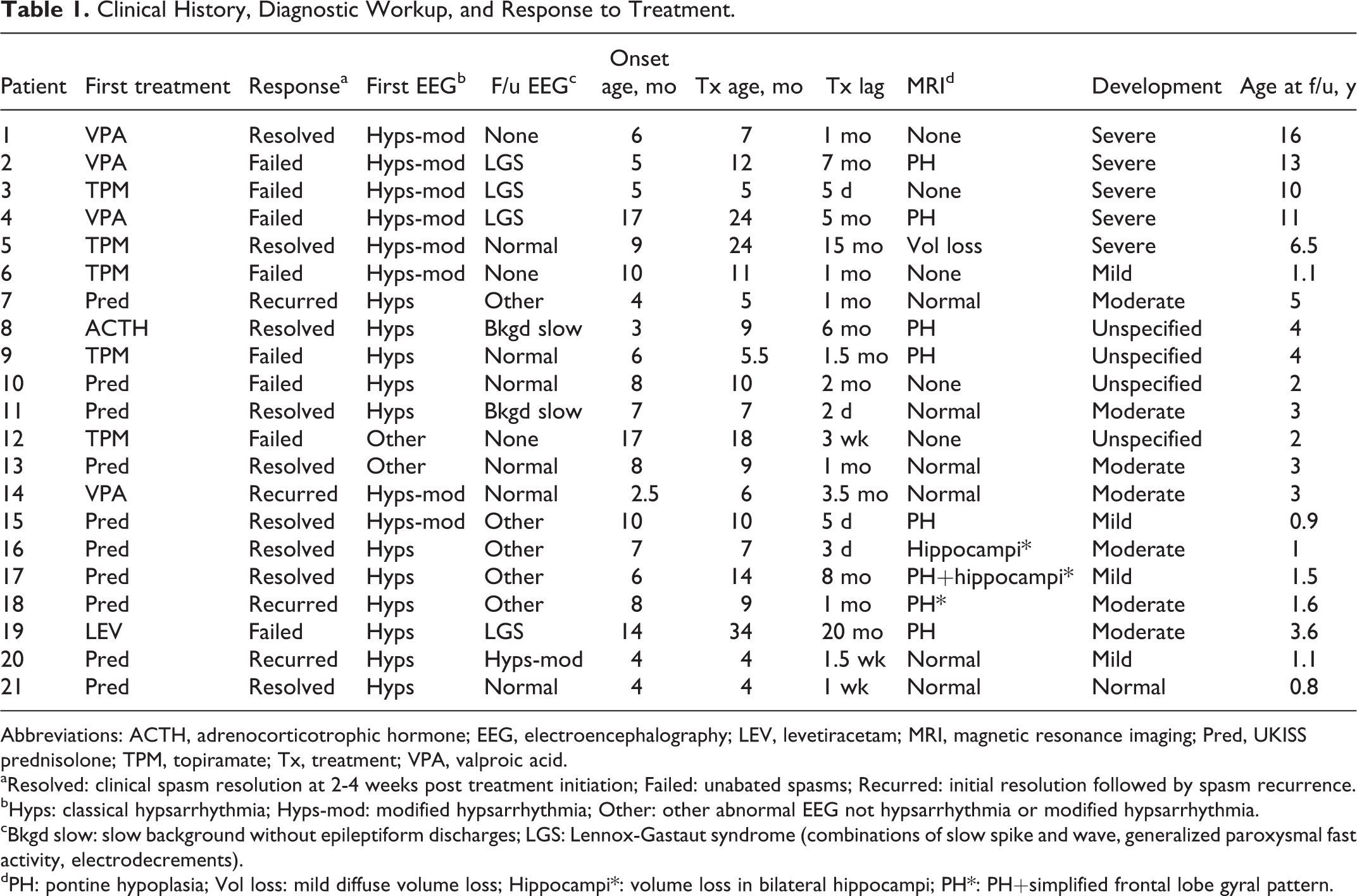
Abbreviations: ACTH, adrenocorticotrophic hormone; EEG, electroencephalography; LEV, levetiracetam; MRI, magnetic resonance imaging; Pred, UKISS prednisolone; TPM, topiramate; Tx, treatment; VPA, valproic acid.
aResolved: clinical spasm resolution at 2-4 weeks post treatment initiation; Failed: unabated spasms; Recurred: initial resolution followed by spasm recurrence.
bHyps: classical hypsarrhythmia; Hyps-mod: modified hypsarrhythmia; Other: other abnormal EEG not hypsarrhythmia or modified hypsarrhythmia.
cBkgd slow: slow background without epileptiform discharges; LGS: Lennox-Gastaut syndrome (combinations of slow spike and wave, generalized paroxysmal fast activity, electrodecrements).
dPH: pontine hypoplasia; Vol loss: mild diffuse volume loss; Hippocampi*: volume loss in bilateral hippocampi; PH*: PH+simplified frontal lobe gyral pattern.
Neuroimaging
Sixteen of the 21 patients had brain MRIs: 38% (n = 6/16) were normal, 50% (8/16) had hypoplasia of the brainstem and/or ventral pons, with 2 of these 8 demonstrating additional findings of either bilateral hippocampal volume loss or simplified gyral pattern in bilateral frontal lobes, and the final 2 studies exhibited either diffuse volume loss or bilateral hippocampal volume loss. Of the 5 patients without brain MRI scans, 1 had a normal computed tomography scan and the other 4 did not have imaging at our institution.
EEG Characteristics
At the time of initial EEG, (median age 9 months, interquartile range 5.75-13 months), 11 (52%; 11/21) had classic hypsarrhythmia, 8 (38%; 8/21) had modified hypsarrhythmia, whereas the remaining 2 (10%; 2/21) patients’ first EEGs demonstrated some combination of independent multifocal discharges, continuous delta slowing, poor background organization, with 1 having a clinical correlation of West syndrome.
Additionally, all EEGs recorded sleep, but the duration of the diagnostic and follow-up EEGs varied across patients; between 40-minute and >24-hour EEG studies.
Response to Treatments
The timing of clinical spasms remission data varied throughout the 14-year time period, but fell between 2 and 4 weeks after initiation of treatment. Of note, patient 1 was lost to follow-up for many years after initial treatment in hospital and has no follow-up EEG, but re-established care with genetics clinic later in childhood, with clinic documentation noting resolution of spasms with valproic acid therapy. Patient 6 did not follow up with neurology after an initial follow-up visit during which spasms were ongoing and topiramate was increased. For this reason, this patient has no follow-up EEG. The median treatment lag was 1 month (interquartile range 0.25-5.5 months) with a range of 2 days to 20 months after onset of clinical spasms. Most patients in the cohort (57%; 12/21) were treated within 1 month of clinical onset.
Of the 21 patients with trisomy 21 and infantile spasms, 10 patients (48%; 10/21) received a nonstandard first-line ASM (5 with topiramate, 4 with valproic acid, and 1 with levetiracetam). Two of these 10 children (20%) achieved spasm control after first-line treatment: 1 patient treated with valproic acid and 1 patient treated with topiramate. The remaining 8 patients did not respond to first-line therapy (80% failure rate; 8/10). Four of those 8 (50%; 4/8) progressed to Lennox-Gastaut syndrome. As previously mentioned, the patient who responded to valproic acid did not have a repeat EEG. The patient who responded to topiramate had a normal follow-up EEG. The other 8 patients who did not respond to nonstandard first-line therapies had follow-up EEGs with various combinations of generalized slowing, multifocal epileptiform discharges, generalized epileptiform discharges, generalized paroxysmal fast activity, and electrodecremental segments. A single patient who failed valproic acid therapy later responded to prednisolone and had a normal follow-up EEG.
Of the remaining 11 patients who received standard first-line therapy, 1 was treated with ACTH and 10 were treated with prednisolone. Two patients were treated with vigabatrin, but were excluded from analysis because of lack of follow-up at our institution. Nine of the 10 patients (90%) who received prednisolone responded to therapy with clinical spasm resolution. Their clinical characteristics are summarized in Table 2. Six of the 9 responders (60% of prednisolone group; 6/10) achieved clinical spasm resolution without recurrence at the time of last follow-up (median 13 months after treatment, interquartile range 5.5-33 months). The 3 patients whose spasms recurred during tapering of prednisolone continued to have spasms despite treatment with topiramate (2 patients) and ACTH (a third patient). None of the patients who received prednisolone reported serious adverse effects. Anticipated adverse effects of irritability and weight gain occurred frequently and resolved with taper.
First-Line Prednisolone Cohort.a

Abbreviation: IQR, interquartile range.
aFor explanations of abbreviations used, please refer to the notes in Table 1.
In the 6 children who responded to prednisolone without recurrence, 3 patients had normal or moderately slow follow-up EEGs, and the other 3 patients had slow backgrounds with epileptiform discharges in various locations. None of the responders demonstrated continued hypsarrhythmia on post-treatment EEGs. In the 3 other patients treated with prednisolone, initial spasm resolution was followed by recurrence during the course of the steroid taper. Their follow-up EEGs revealed slow backgrounds with epileptiform discharges in various locations. Infantile spasms continued without abatement in 1 patient treated with prednisolone (10%; 1/10). He was subsequently treated with topiramate with resolution of clinical spasms and normalization of follow-up EEG, which was completed 18 months after initial treatment with steroids.
The final patient received ACTH as first-line therapy (9% of the standard therapy group; 1/11) and experienced spasm resolution and normalization of follow-up EEG. There had been no recurrence of spasms or other seizure types at the last follow-up visit at age 4 years and no reported significant adverse effects during hormonal therapy.
Given the small number of patients in each group (resolution vs resolution with recurrence and initial treatment failure combined), a Fisher exact test was performed to compare the standard and nonstandard treatment groups. The result was .0805, which did not reach statistical significance.
Developmental Outcomes
Retrospective developmental delay assessment was performed on all patients in our series using our standardized scale.
Five patients (24%; 5/21) had severe delay at last follow-up (median age 10 years, range 0.66- 16 years). Seven patients (33%; 7/21) had moderate delay (median age 3 years, range 1-5 years). Four patients (19%; 4/21) had mild delay (median age 13 months, range 11-18 months). One patient (5%; 1/21) had normal development at time of last follow-up at age 10 months. Four patients (19%; 4/21) had unspecified delay.
Group Comparison
The ages of spasm onset were similar in the standard group (median 7 months; interquartile range 4-8 months) and the nonstandard group (7.5 months; 5-14 months). Median treatment lag in the standard group was somewhat shorter (1 month; 0.13-2 months) when compared to the nonstandard group (2.5 months; 1-7 months). The combination of hypsarrhythmia and modified hypsarrhythmia versus other EEG results was similar in both groups (standard group: classical and modified hypsarrhythmia in 10, abnormal without hypsarrhythmia in 1; nonstandard group: classical and modified hypsarrhythmia in 9, abnormal without hypsarrhythmia in 1).
The MRI results in the 2 groups were similar with regard to incidence of pontine hypoplasia (standard 36%;4/11 vs nonstandard 40%; 4/10) and nonspecific abnormalities (standard: 9%;1/11 vs nonstandard 10%;1/10). Normal MRI results were documented in 45% (5/11) of the standard treatment group and only 10% (1/10) of the nonstandard treatment group. Conversely, only 9% (1/11) of the patients who received standard first-line therapy did not have an MRI, but 40% (4/10) of the patients in the nonstandard group had no MRI.
Four patients were diagnosed with Lennox-Gastaut syndrome in the nonstandard group, whereas no patient had this diagnosis in the standard treatment group. No patients in the standard group had severe developmental delay at the last follow-up visit, whereas 5 of the children with nonstandard first-line treatments had severe delay at last clinic visit. This comparison is summarized in Table 3.
Group Comparison.a
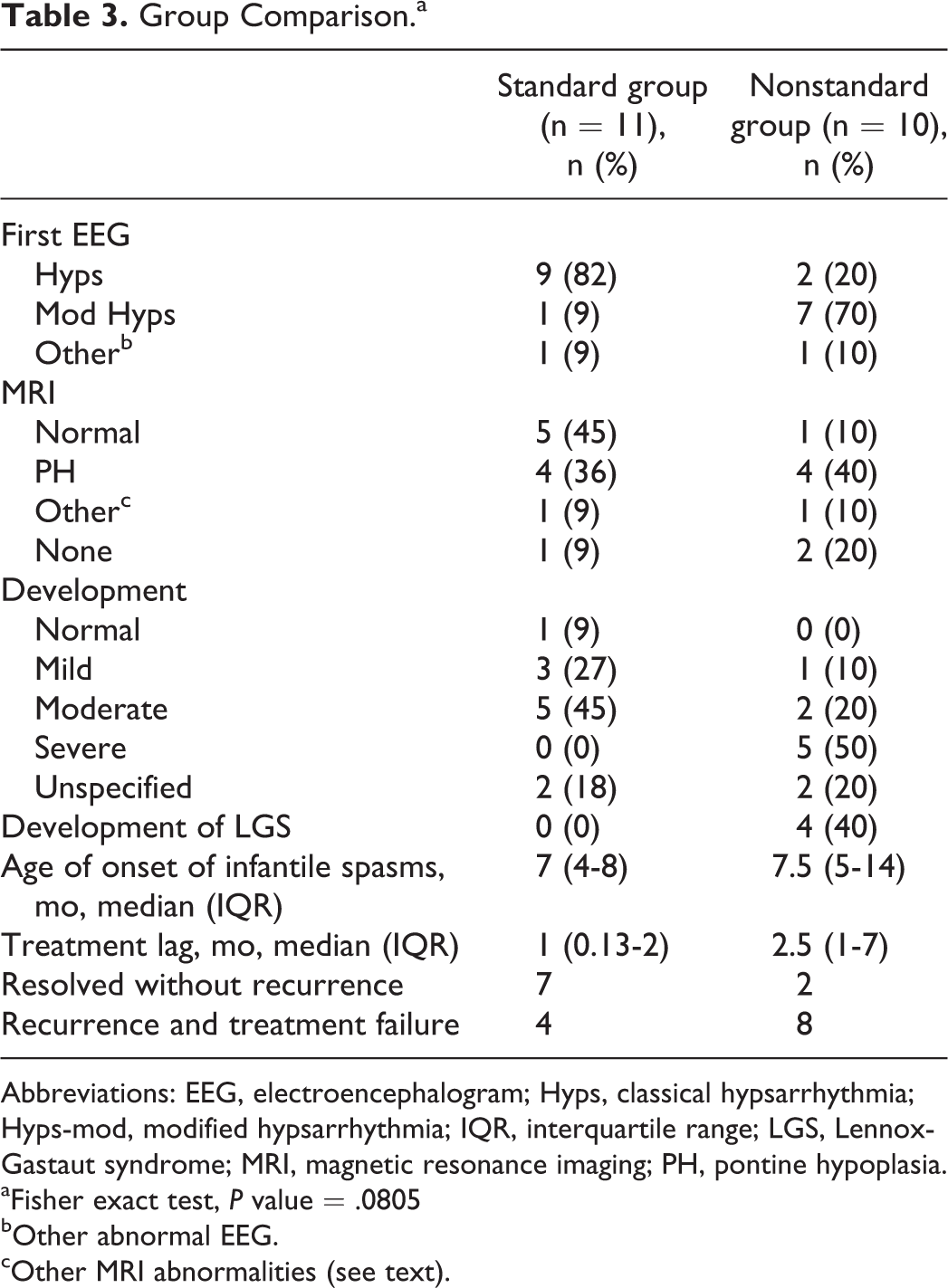
Abbreviations: EEG, electroencephalogram; Hyps, classical hypsarrhythmia; Hyps-mod, modified hypsarrhythmia; IQR, interquartile range; LGS, Lennox-Gastaut syndrome; MRI, magnetic resonance imaging; PH, pontine hypoplasia.
aFisher exact test, P value = .0805
bOther abnormal EEG.
cOther MRI abnormalities (see text).
Discussion
In addition to being the largest series of children with trisomy 21 and infantile spasms to our knowledge in the literature, this study is the first to report a series in the same population with a significant proportion of patients who received UKISS-protocol prednisolone since the results of that study were published in 2004. 7 Sixty percent of our patients who were treated with prednisolone first line experienced resolution of infantile spasms and hypsarrhythmia without subsequent recurrence and without significant adverse effects. This suggests that prednisolone is an effective and well-tolerated first-line therapy in children with infantile spasms due to trisomy 21. Other studies have investigated first-line treatment with ACTH in children with trisomy 21 and infantile spasms, 9,10 but have not had a significant proportion of children who were treated with UKISS protocol prednisolone.
Conversely, the lack of response to nonstandard first-line treatments also bears mention. Only 2 of the 10 children treated first-line with nonstandard therapies experienced remission. This responder rate (20%) is similar to the nonstandard therapy response rates in a recent large series through the Pediatric Epilepsy Research Consortium, 16 in which 3 of 32 (9%) responded. Of note, the more recently treated patients in the cohort were more likely to receive standard first-line treatments. Additionally, the median treatment lag also differed. The nonstandard group’s median lag is 2.5 months (interquartile range of 1-7 months), whereas the standard group has a median of 1 month (interquartile range of 0.13-2 months). One study 17 showed that treatment lags of greater than 2 months are associated with worse clinical seizure control and worse long-term development. Our standard treatment group median lag and interquartile range fall within that statistically significant delineation, and our developmental findings are consistent with the trend noted in that group as well. Differences in treatment lag may not be the only factor related to long-term seizure control or developmental outcomes; there are patients such as the patient in this series who failed valproic acid and later responded to UKISS-protocol prednisolone who fail multiple therapies and later experience resolution with first-line therapies.
Additionally, the only children with Lennox-Gastaut syndrome were those who were treated with nonstandard therapies. This correlation is tempered, however, by the knowledge that progression to Lennox-Gastaut syndrome requires observation time, and many of the children in the standard therapies treatment group are more recently treated, and may go on to relapse in future or to develop new seizure types, and may ultimately progress to Lennox-Gastaut syndrome. Evaluating this trend further would require a longer observation period.
Development
Although there remain no developmental scales validated specifically for children with trisomy 21, 13 –15 many clinics apply validated developmental scales used for typically developing children in this patient population. 18 Multiple studies 9,10 have discussed a need to assess the effect on development of early, appropriate infantile spasms treatment in these children. A 2013 case series attempted to apply a validated developmental tool by calling each patient family to administer a Vineland Adaptive Behavioral Scale, Modified Checklist for Autism in Toddlers, or Social Communication Questionnaire. 10 More recently, a 2016 collaborative effort between 22 pediatric tertiary care centers applied a framework of 3 generalized developmental groupings similar to the one employed in our study. 16
Although the scale used in our study lacks validation, it was a useful tool to evaluate trends of developmental delay. Four of the 5 patients who were noted to have severe delay were older at last follow-up (6.5-16 years old). The correlation between nonstandard first-line therapy and more severe developmental delay is interesting, but the practice of treating children with trisomy 21 and infantile spasms with nonstandard therapies such as valproic acid and topiramate was much more common at our institution in the earlier years of our cohort. Additionally, the scale applied in our study is based on the developmental quotient, which more clearly shows delay at older ages. Older children are more likely to be identified as severely delayed in development because the differences in their abilities compared with peers will be more obvious, and skills such as walking, talking, and self-feeding are well documented in the medical record. Conversely, the patients with mild delay were all the most recently treated patients in the series, so they may have a more severe delay than is suggested by their mild delay designation based on our developmental delay categories. It is possible that further clinical follow-up may identify larger gaps in development when compared to typically developing peers. Overall, our developmental data suggest that further studies should employ a prospective, validated developmental scale in order to examine the developmental effects of early, aggressive treatment of infantile spasms in children with trisomy 21.
Neuroimaging
Eight of the patients who had an MRI available for review had pontine hypoplasia; this has previously been described in multiple cohorts of patients with trisomy 21. 19,20 Though the series of patients with trisomy 21, pontine hypoplasia, and infantile spasms is small in our cohort, interestingly, only those with additional structural abnormalities did not respond to hormonal therapies. Children with trisomy 21 frequently have an abbreviated infantile spasms workup because their etiology is felt to be their congenital genetic abnormality. Further study regarding the frequency of midbrain hypoplasia in this population, as well as response to first-line therapies in children with trisomy 21, infantile spasms, and midbrain hypoplasia would add to the current knowledge about the clinical significance of this finding as it pertains to epilepsy treatment response.
Limitations
Despite being the largest series of trisomy 21 and infantile spasms patients in the literature to our knowledge, our study is limited in number and lacks statistical power to show differences in efficacy of first-line therapies. Additionally, although there is a significant proportion of patients who were treated with prednisolone, there is only 1 patient who was treated first-line with ACTH, another reason this work cannot make a comparison between these 2 therapies. The treating clinician’s choice of first-line medication may have been influenced by a number of undocumented factors such as degree of prior developmental delay, parental preference, cost concerns, or personal experience with infantile spasms cessation in trisomy 21. As with other retrospective analyses, our study does not have standardized follow-up EEG timing or length, and dosing for the nonhormonal therapies varied by neurologist.
However, all patients treated with prednisolone received a standardized dosing regimen consistent with the UKISS protocol. Though some studies use a more strict electroclinical remission criteria, such as the 2013 series in which a 24-hour video-EEG confirming resolution of hypsarrhythmia was obtained within 3 months of therapy, 10 many centers do not have access to the EEG resources to make this standard practice. Our study uses clinical resolution of spasms and electrographic resolution of hypsarrhythmia as its treatment endpoint. An ideal study would be a prospective, randomized trial in which children with trisomy 21 who developed infantile spasms could be allocated to receive either high-dose ACTH, high-dose prednisolone, or vigabatrin and have follow-up 24-hour video EEGs with tracings interpreted by a single, blinded reviewer to confirm abolition of hypsarrhythmia, and with routine clinical follow-up up to years to surveil for late relapse. Despite these limitations, our study adds helpful information about the safety and tolerability of high-dose prednisolone in this cohort.
Conclusions
This cohort of children with trisomy 21 and infantile spasms is the first to have a significant proportion of patients who received prednisolone according to the United Kingdom Infantile Spasms Study protocol rather than a majority receiving ACTH. Although it is underpowered to show a difference between prednisolone and ACTH, it does demonstrate that prednisolone is a safe and well-tolerated treatment in this group.
Footnotes
Acknowledgments
The authors wish to thank the patients and families who share their experiences with their neurologists, as well as the electroencephalography (EEG) technologists at our institution who care for these patients as well. Thanks to Rachel S. Rogers, MS, who shared her Stata license and expertise in biostatistics for this work.
Author Contributions
DA completed the chart review and composed the initial draft and subsequent edits of this manuscript. RS performed the electroencephalography (EEG) database query, contributed to editing drafts of this report, and served as a mentor for the project.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Ethical Approval
This study was approved by the Institutional Review Board of UT Southwestern Medical Center.