
Abstract
The ring-finger protein 213 (RNF213) gene is a major susceptibility gene for moyamoya disease. The homozygote of the p.R4810K variant on RNF213 exhibits an early onset age and severe form of moyamoya disease. We report 4 unrelated pediatric moyamoya disease cases with the homozygous p.R4810K variant and the long-term surgical outcomes. Published reports on surgical outcome of moyamoya disease case with the homozygous p.R4810K variant were reviewed. Cerebral angiography revealed classic angiographic findings of moyamoya disease in 7 hemispheres of the 4 children. All patients underwent successful indirect revascularization. Abundant collateral blood flow from the external carotid arteries to the internal carotid arteries was observed in all bypass procedures by angiography. Improvements in symptoms and cerebral blood volume were observed in all patients at long-term follow-up. This report is the first case series in the literature on the surgical management of these patients. These cases highlight the effectiveness of indirect revascularization for moyamoya disease patients with the homozygous p.R4810K variant. Early diagnosis and treatment are crucial to avoid irreversible neurologic deficits in these patients.
Moyamoya disease is an idiopathic cerebral vasculopathy that is characterized by a progressive stenosis of the terminal portion of the internal carotid arteries and the development of a network of abnormal collateral vessels. 1 It is the most common pediatric cerebrovascular disease in East Asia. Revascularization surgery for symptomatic moyamoya disease is the standard treatment for preventing further stroke and decreasing cognitive deficits. 1 –3
The ring-finger protein 213 (RNF213) gene was identified recently as a major susceptibility gene for moyamoya disease. 4,5 A founder variant, p.R4810K (c.14429G>A; rs112735431), which was previously annotated as p.R4859K (c.14576G>A; GenBank: XM_005257545.3 and XP_005257602.2), was significantly associated with the risk of moyamoya disease in a series of studies in East Asian populations. 4 –7 The homozygote of the c.14576G>A variant (p.R4810K) shows an early onset age and rapid disease progression, which results in significant neurologic deficits with a severe and wide distribution of vasculopathy. 8 –10 The natural history and clinical feature of pediatric moyamoya disease patients with homozygous p.R4810K variant is unclear. Moreover, the long-term surgical outcome for homozygotes of the p.R4810K variant is not reported.
We report 4 unrelated pediatric moyamoya disease cases with the homozygous p.R4810K variant and their long-term surgical outcomes.
Patients and Methods
Moyamoya disease cases from inpatient and outpatient departments of the Beijing Tiantan Hospital from June 2012 to May 2016 were reviewed. The following inclusion criteria were used: (1) pediatric patients (age <15 years old) diagnosed in accordance with the Japanese moyamoya disease diagnosis guidelines 2 ; (2) patients with confirmed homozygous p.R4810K variant of RNF213; (3) patients with complete clinical and radiologic data available.
Clinical and radiologic information were obtained from clinical chart review. Surgical procedures included encephaloduroarteriosynangiosis, multiple burr hole, and combined procedure. Encephaloduroarteriosynangiosis is usually the first choice for most hemispheres, in which the posterior branch of superficial temporal artery with its surrounding galeal cuff is sutured to the arachnoid. For patients with chronic and massive cerebral ischemia (usually ≥3 lobes in a hemisphere), multiple burr hole or a combined procedure that perfuses more cerebral areas will be adopted. During the burr hole procedure, a flap of periosteum is placed into the burr hole, making direct contact with the exposed cortex. Long-term outcome after discharge was ascertained during clinical visits. Genetic testing for the p.R4810K variant of RNF213 and magnetic resonance angiography examination were suggested tests for patient’s family members, including unaffected members. The following primers were designed: RNF213-4810F (rs112735431): 5′-GCCCTCCATTTCTAGCACAC-3′; RNF213-4810 R: 5′-AGCTGTGGCGAAAGCTTCTA-3′. Our previous studies describe the method in detail. 6,11 The Ethics Committee of Beijing Tiantan Hospital, Capital Medical University, Beijing, China, approved the study.
Review of the Pertinent Literature
Published reports on surgical outcome of pediatric moyamoya disease cases with the homozygous p.R4810 K variant were reviewed. The MEDLINE database was searched using the key words RNF213, p.R4810K, c.14576G>A, and moyamoya. Reports that included clear clinical and surgical outcome information of pediatric moyamoya disease patients with the p.R4810K or c.14576G>A (rs112735431) variant were reviewed for comparison with our observations.
Results
Patient 1 and Family 1
A 10-year-old boy presented with right-sided hemiparesis and generalized epilepsy diagnosed at 3 years of age. He had seizures every 2 to 4 weeks despite the oral administration of oxcarbazepine. A magnetic resonance image (MRI) revealed a left deep watershed zone ischemic infarction. Digital subtraction angiography indicated stenosis-occlusive lesions of the bilateral middle cerebral artery and the development of moyamoya vessels (Figure 1). Computed tomographic (CT) perfusion revealed reduced cerebral blood flow and a prolonged time to peak in the bilateral hemispheres. The patient underwent staged bilateral encephaloduroarteriosynangiosis separated by 6 months. He recovered well. Follow-up angiography revealed abundant filling of the middle cerebral artery regions by the external carotid arteries, bilaterally, and progression of the left posterior cerebral artery. Cerebral blood flow and right-sided hemiparesis improved, and the frequency of seizures decreased as of the last follow-up at 2 years.
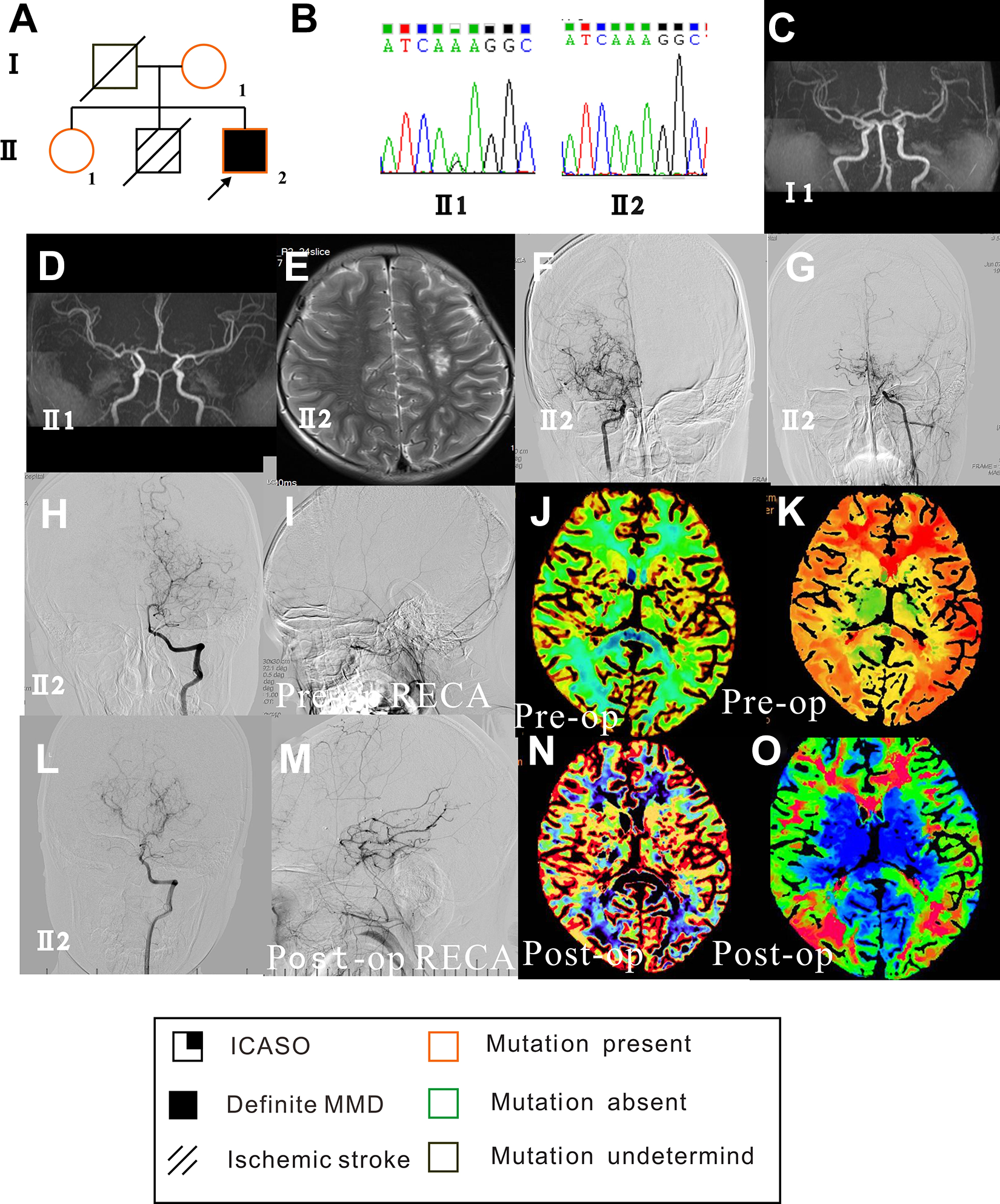
Genetic background, clinical feature, and surgical outcome of case 1. (A) Family pedigree and disease and mutation statuses are indicated in the figure key. The index case (Ⅱ 2) has the homozygous p.R4810 K variant, and the mother (Ⅰ 1) and sister (Ⅱ 1) have the heterozygous (G/A) variant. (B) Sanger sequencing showing (left) the heterozygous variant (G/A) of the sister (Ⅱ 1) and (right) the homozygous variant (A/A) of case 1 (Ⅱ 2). (C and D) Magnetic resonance angiography showing the normal mother (Ⅰ 2) and sister (Ⅱ 1) of case 1. (E) MRI showing the left deep watershed zone ischemic infarction of case 1 (Ⅱ 2). (F, G, H, and I) Preoperative angiography showing the bilateral development of moyamoya vessels. The posterior cerebral artery was normal, and right external carotid artery was not filling the internal carotid artery. (J and K) Preoperative CT perfusion showing reduced cerebral blood flow (J) and prolonged time to peak in bilateral hemispheres (K). (L and M) Postoperative angiography showing a progressing of the left posterior cerebral artery (L) and sufficient filling of the middle cerebral artery by the right external carotid artery (M). (N and O) Postoperative CT perfusion showing improved cerebral blood flow (N) and prolonged time to peak in bilateral hemispheres (O). Figures 1 to 4 share the same figure keys. Disease and mutation status are indicated in the figure key. Circles represent women, and squares represent men. Arrowheads indicate the proband in the family. A diagonal line through a circle or square indicates that the individual is deceased. Each disease is coded and correlates with the figure key. (CT, computed tomography.)
The homozygous p.R4810K variant in RNF213 was identified in the boy, and the heterozygous variant was detected in his mother and sister. There was no steno-occlusive lesion identified in the 2 heterozygous variants. His brother suffered a stroke in his childhood, and his father and brother died sudden unexpected deaths.
Patient 2 and Family 2
A 7-year-old boy presented with left-sided weakness associated with transient ischemic attack. His mother was diagnosed with moyamoya disease at 43 years of age due to infarction onset. An MRI revealed cerebral infarction of the right temporal and occipital lobes. Digital subtraction angiography indicated moyamoya vessels on the right internal carotid artery and a mild narrowing of the left proximal middle cerebral artery (Figure 2). CT perfusion revealed reduced cerebral blood flow in the right hemisphere. The patient underwent right encephaloduroarteriosynangiosis surgery and tolerated the procedures well. Follow-up angiography revealed abundant filling of the right middle cerebral artery region by the external carotid artery. Left-sided weakness improved as of the last follow-up at 6 years. No progression was observed on the left middle cerebral artery. He did not experience further strokes or new neurologic symptoms.
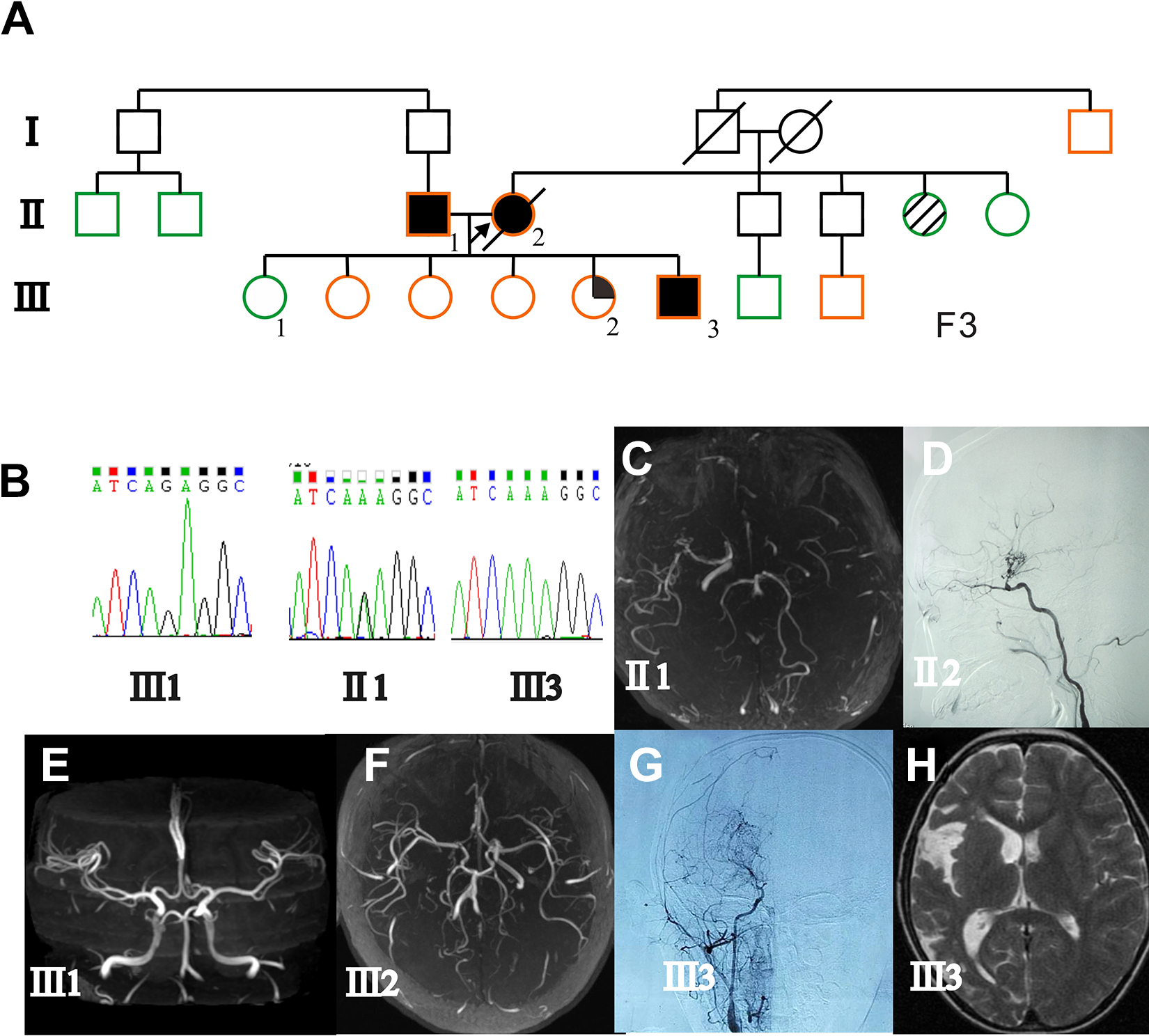
Genetic background and clinical feature of case 2. (A) Family pedigree. Case 2 (Ⅲ 3) has the homozygous p.R4810K variant, and his father (Ⅱ 1), mother (Ⅱ 2), and some relatives have the heterozygous (G/A) variant. (B) Sanger sequencing showing a normal sequence of his oldest sister (Ⅲ 1), a heterozygous variant (G/A) of his father (Ⅱ 1), and the homozygous variant (A/A) of case 2 (Ⅲ 3). (C) Magnetic resonance angiography showing occlusion of the left proximal internal carotid artery, moyamoya changes, and a steno-occlusive lesion of right middle cerebral artery in the father (Ⅱ 1). (D) DSA showing a steno-occlusive lesion of the right middle cerebral artery and development of moyamoya vessels in the mother (Ⅱ 2). (E) Magnetic resonance angiography showing the normal oldest sister. (F) Magnetic resonance angiography showing occlusion of the right proximal internal carotid artery and stenosis of the right anterior cerebral artery of the youngest sister (Ⅲ 2). (G) DSA showing a steno-occlusive lesion of the right middle cerebral artery and development of moyamoya vessels in case 2 (Ⅲ 3). (H) T2-weighted MRI showing infarctions of the right temporal and occipital lobes of case 2 (Ⅲ 3). (DSA, digital subtraction angiography; MRI, magnetic resonance imaging.)
Sanger sequencing revealed a homozygous p.R4810K variant in the boy. The heterozygous variant was detected in his father, his mother, and extended family members (Figure 2). Magnetic resonance angiography screening of his family members identified an moyamoya disease patient with mild symptom, and his youngest sister as an asymptomatic intracranial major artery stenosis/occlusion patient. His mother died of stroke complication at the last follow-up.
Patient 3 and Family 3
A 3-year-old boy presented with right-sided hemiparesis, aphasia, and bilateral visual impairment. He had a history of generalized convulsions and was diagnosed with cerebral infarction at 2 years old. CT and MRI revealed multiple cerebral infarction of bilateral frontal, temporal, parietal, and occipital lobes. Digital subtraction angiography indicated moyamoya vessels on the bilateral anterior and posterior circulation (Figure 3). Arterial spin-labeling MRI revealed reduced cerebral blood flow in the bilateral hemispheres. He underwent encephaloduroarteriosynangiosis plus multiple–burr hole procedure on the left hemisphere. Asymptomatic cerebral infarction was observed in the surgical hemisphere postoperatively. Another revascularization surgery, multiple–burr hole procedure, was performed on the right hemisphere. Subdural hematoma was a postoperative complication after the second surgery, and it was treated conservatively. Follow-up angiography revealed abundant filling of the middle cerebral artery regions by the external carotid arteries, bilaterally. Right-sided hemiparesis and aphasia improved and visual impairment persisted as of the last follow-up at 2 years. Mild hemiparesis remained on both sides. He cannot perform basic activities of daily living independently.
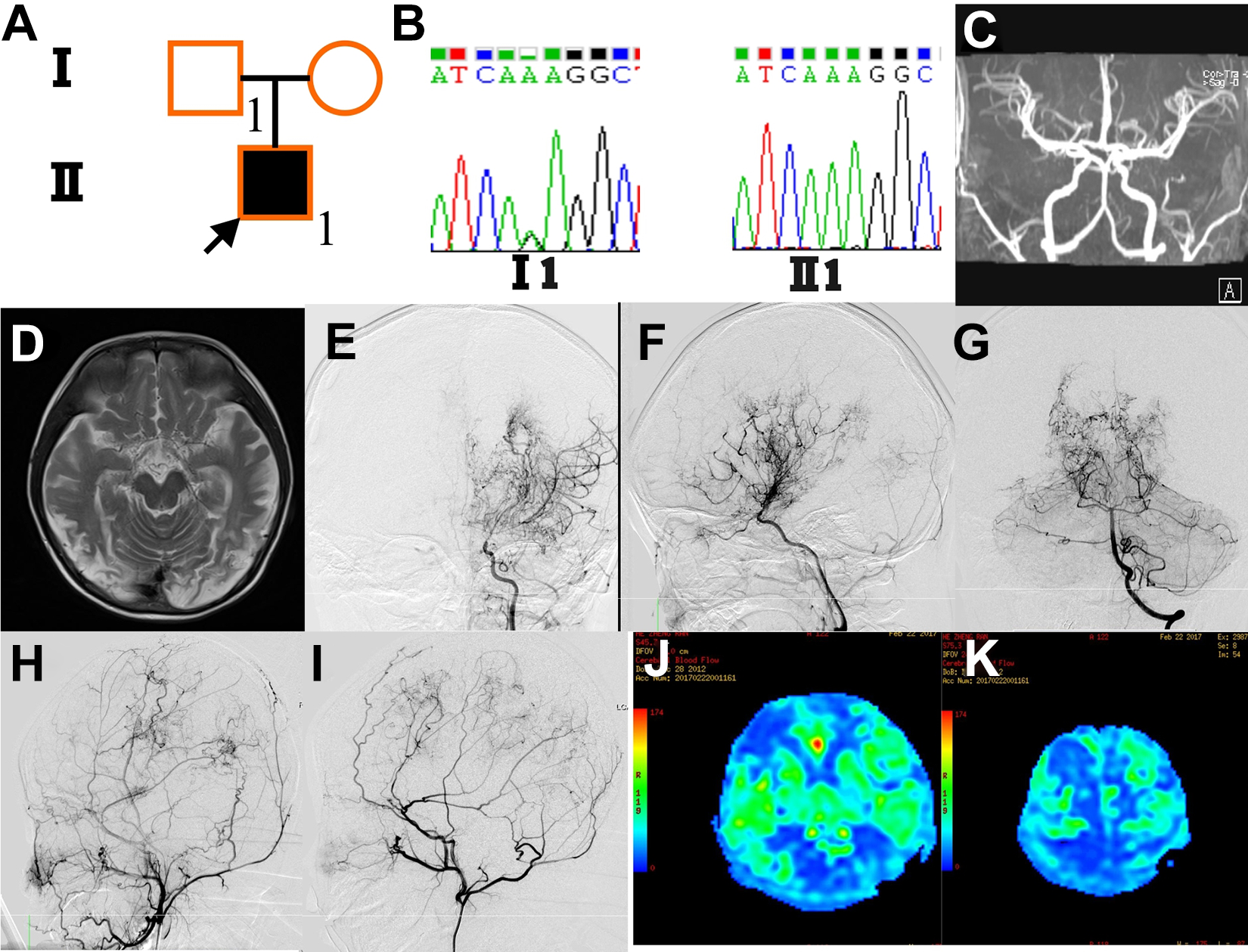
Genetic background, clinical feature, and surgical outcome of case 3. (A) Family 3 pedigree. Case 3 (Ⅱ 1) has the homozygous p.R4810K variant, and the father (Ⅰ 1) and mother have the heterozygous (G/A) variant. (B) Sanger sequencing showing a heterozygous variant (G/A) of his father (Ⅰ 1) and the homozygous variant (A/A) of case 3 (Ⅱ 1). (C) Magnetic resonance angiography showing the normal father. (D) T2-weighted MRI showing multiple cerebral infarctions of bilateral frontal, temporal, parietal, and occipital lobes in case 3 (Ⅱ 1). (E, F, and G) Preoperative angiography showing left development of moyamoya vessels on the left anterior (E, F) and posterior circulation (G) of case 3. (H and I) Angiography after surgical treatment of the patient showing abundant filling of bilateral middle cerebral artery region by the external carotid artery. (J and K) Arterial spin-labeling MRIs show improved cerebral blood flow in the bilateral hemispheres. (MRI, magnetic resonance imaging.)
The genetic sequencing indicated the boy as a homozygote of p.R4810K variant, and the heterozygous variant was detected in his mother and father. No steno-occlusive lesions were identified in his parents.
Patient 4
A 5-year-old girl presented with left-sided hemiparesis and aphasia. She had a first history of recurrent left-sided weakness and was diagnosed with cerebral infarction at age 3 years. CT scan revealed multiple cerebral infarctions of the right occipital and bilateral frontal lobes and cerebral atrophy. Digital subtraction angiography indicated moyamoya vessels on the bilateral internal carotid artery and left posterior cerebral artery (Figure 4). Single-photon emission CT revealed a marked reduction of cerebral blood flow, particularly in the frontal and temporal lobes. The patient underwent a multiple–burr hole procedure on the right hemisphere and encephaloduroarteriosynangiosis plus a multiple–burr hole procedure on the left hemisphere separated by 6 months. She tolerated the procedures well. Follow-up angiography revealed abundant filling of the middle cerebral artery regions by the external carotid arteries, bilaterally. The patient’s symptoms were improved at the 5-year follow-up, and no new strokes or perfusion defects occurred in the follow-up period. The homozygous p.R4810K variant was identified.
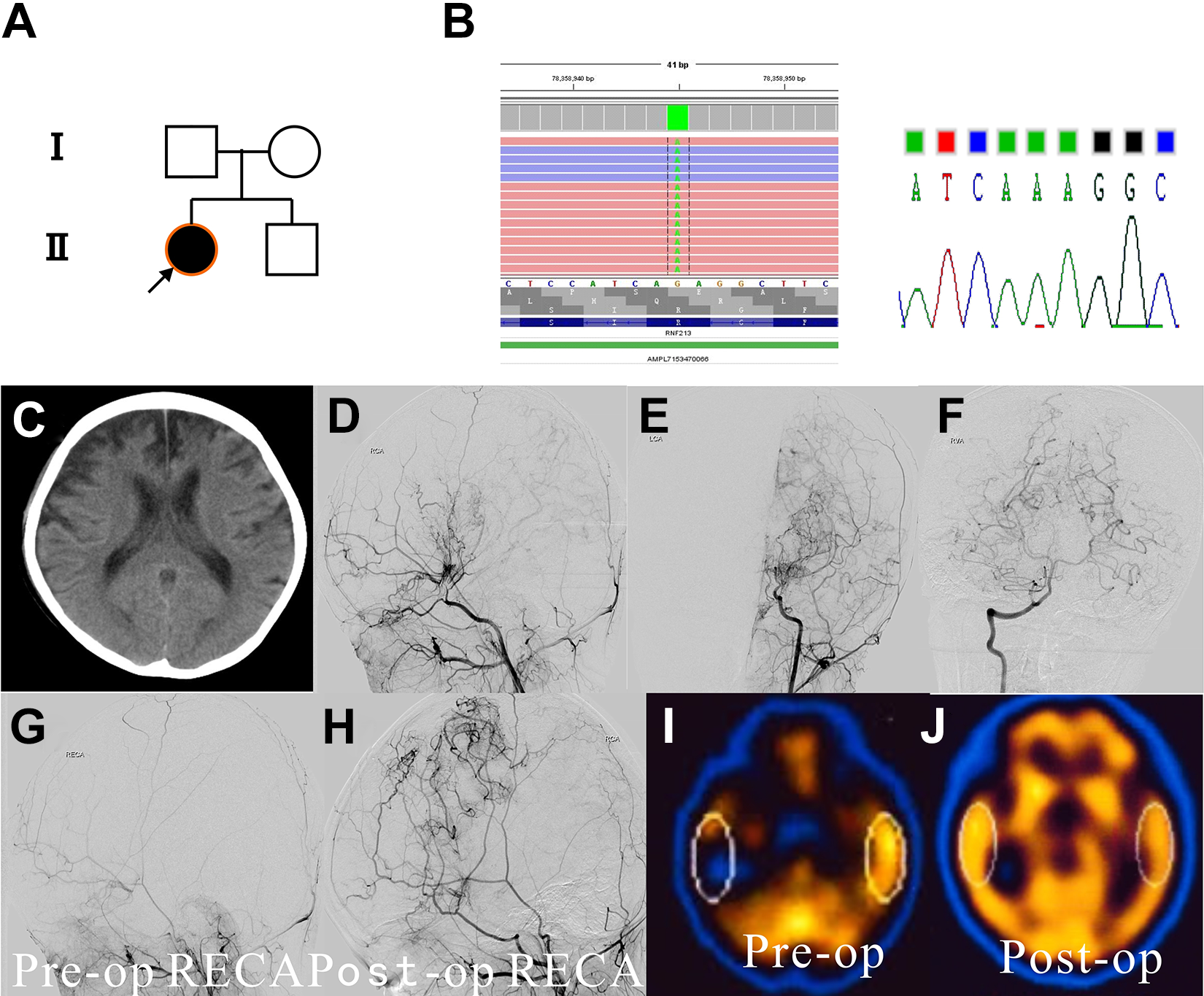
Genetic background, clinical feature, and surgical outcome of case 4. (A) Family 4 pedigree. Case 4 has the homozygous p.R4810K variant. (B) Next-generation sequencing and Sanger sequencing results. (Left) Next-generation sequencing results of the patient using IGV viewer. The RNF213 variant, p.R4810K, was detected as demarcated by dotted lines. (Right) Sanger sequencing showing the homozygous variant (A/A) of case 4. (C) CT image showing multiple cerebral infarctions of the right occipital lobe and bilateral frontal lobes and cerebral atrophy in case 4. (D and E) Preoperative angiography showing bilateral development of moyamoya vessels in case 4. (F) Preoperative angiography showing normal posterior cerebral arteries in case 4. (G and H) Angiography prior to (G) and 6 months after surgical treatment (H) of the patient showing abundant filling of the middle cerebral artery region by the external carotid artery. (I and J) Single-photon emission CT images before (left) and after (right) surgery showing improved cerebral blood flow in the bilateral hemispheres, particularly in the frontal and temporal lobes. (CT, computed tomography; IGV, Integrative Genomics Viewer (Broad Institute and the Regents of the University of California), Broad Institute)
Review of the Pertinent Literature
The surgical outcome of 1 moyamoya disease case with the homozygous p.R4810K variant was identified. The patient had a first history of transient ischemic attack associated with left-sided weakness at age 2. 12 Bilateral encephaloduroarteriosynangiosis and frontal omental transplantation were performed at age 2 and 3, respectively. Later, transient ischemic attacks associated with visual disturbance and right-sided weakness prompted bilateral occipital gracilis muscle transplantations in occipital lobes and subcutaneous tissue graft transplantation in the left parietal lobe. The transient ischemic attacks subsequently decreased. Currently, a mild to moderate hemiparesis is evident on both sides. 12 We denoted this patient as patient 5. Table 1 summarizes the clinical features and surgical outcomes of the 5 homozygotes.
Clinical Features and Surgical Outcomes of Patients With Moyamoya Disease and the Homozygous RNF213 p.R4810K Variant in the Current Study and Others Reported in the Literature.

Abbreviations: EDAS, encephaloduroarteriosynangiosis; mRS, modified Rankin Scale; NA, not available; PCA, posterior cerebral artery, STA, superficial temporal artery; STV, superficial temporal vein.
Discussion
RNF213 is the major susceptibility gene for moyamoya disease in East Asia. 4 –7 The prevalence of homozygosity for the RNF213 p.R4810K variant is estimated to be 7% to 8% of all patients with moyamoya disease in Japan and Korea. 7,13 The incidence rate of moyamoya disease in homozygotes is >78%, but the penetrance rate of moyamoya disease in heterozygotes is low, with 0.33% to 0.67% (1/150-300). 7,13 Our previous study identified 2 pediatric homozygotes among 255 Chinese moyamoya disease patients (0.78%). 6 Miyatake et al 12 reported a family with sibling pediatric moyamoya disease cases who were homozygous and heterozygous for the RNF213 variant, which demonstrated the dosage effect of the variant on clinical phenotype. Four homozygotes manifested as childhood onset moyamoya disease in this study. Magnetic resonance angiography and sanger sequencing were performed in 18 family members of the 4 families. Two moyamoya disease cases and an asymptomatic intracranial major artery stenosis/occlusion case were identified in 12 family numbers, who harbored the heterozygous variant. No moyamoya disease or intracranial major artery stenosis/occlusion cases were identified among the other 6 wildtype family members. Our results support the gene dosage effects of RNF213 R4810K variants on clinical phenotype.
The clinical manifestation of all homozygotes in our study was ischemic symptoms. Early age onset, cerebral infarction, and posterior cerebral artery involvement were identified in 3 of 4 cases. These findings are consistent with those of a previous study in which the homozygous variant of RNF213 predicted a severe form of moyamoya disease. Pulmonary artery hypertension and hypertension secondary to peripheral pulmonary artery stenosis and renal arteries stenosis as co-occurrence symptoms were also reported in homozygotes of the p.R4810K variant in 6 cases. 8,10 Peripheral pulmonary artery stenosis typically developed in adolescence to young adulthood and progressed slowly. 10 However, homozygotes in our study were children, which may explain the negative findings of pulmonary artery hypertension symptoms in our cases.
Suzuki stages 3 and 4, which represent the classic angiographic findings of moyamoya disease, and the development of pathologic collateral vessels was observed in 7 hemispheres in our study preoperatively. Collateral vessels from the external carotid artery to the internal carotid artery developed soon after indirect revascularization procedures in all homozygotes. These phenomena suggest the potential influence of the RNF213 variant on endothelial dysfunction in a gene dose-dependent manner. This result is consistent with that of a previous study in which inhibition of RNF213 expression induced an abnormal development of ocular arteries in zebrafish. 4 Recent in vitro molecular studies revealed that the p.R4810K variant induced endothelial dysfunction, which may ultimately lead to arterial stenosis and the development of moyamoya vessels. 14 Further research is needed to elucidate the role of the homozygous p.R4810K variant of RNF213 in the course of developing collateral vessels.
Severe-disease phenotype and rapid disease progression were reported in moyamoya disease patients with the homozygous p.R4810K variant. 7,12,13 In this study, good development of collaterals from the external carotid artery to the internal carotid artery and improvements in symptoms and cerebral perfusion were observed in all children. Cerebral plasticity may provide a theoretical basis for this result. 15 Postoperative recovery of the central nervous system is better in children than in adults because the developmental brain is more plastic than the mature brain. 16 –18 A single direct bypass could provide a large volume of blood flow to ischemic tissue relying on the integrity of the vascular network. For patients with widely damaged cerebral vasculature, a direct bypass remains isolated to a single ischemic area without enough canals to perfuse extensive brain areas. 19 Compared to direct bypass, encephaloduroarteriosynangiosis and multiple–burr hole procedure, which do not rely on vascular integrity, will create new vascular network to perfuse the ischemic cortex and repair the damaged links and nodes of the vascular system. Our study observed an increased blood transfusion area and recovery of the entire vascular network in moyamoya disease patients at late stage. Therefore, we recommend enlarged encephaloduroarteriosynangiosis plus multiple–burr hole surgery or multiple–burr hole procedure for these patients.
Multiple infarctions in the developing brain, especially infarctions that affect both hemispheres, lead to poor clinical outcomes, intellectual impairment, and developmental delays in homozygotes. 7,12,13 Delayed surgical treatment was observed in 3 of 4 patients in this study. Therefore, early diagnosis and treatment are crucial for avoiding irreversible neurologic deficits. Early diagnosis in Matsuda and colleagues’ 20 study and our previous work was successfully obtained via screening for the RNF213 variant and magnetic resonance angiography examinations in moyamoya disease family members. 21,12 Noninvasive prenatal gene detection and genetic counseling may ultimately decrease cumulative disability for homozygotes of the p.R4810K variant.
Limitations
The findings of this work should be considered in the context of several limitations. First, this study was limited by sample size (n = 4) owing to the extreme low prevalence of moyamoya disease individuals with the homozygous p.R4810K variant. 7,12,13 Second, this work was a retrospective study and the surgical strategy for 7 hemispheres varied, including multiple burr hole, encephaloduroarteriosynangiosis, and burr hole plus encephaloduroarteriosynangiosis.
Conclusions
Moyamoya disease cases with the homozygous p.R4810K variant manifest as a severe-disease phenotype. Indirect revascularization is effective in these patients. Early surgical intervention should be considered to prevent symptom progression later in life. Furthermore, a large prospective trial is needed to evaluate the individualized revascularization strategy for homozygous patients.
Footnotes
Acknowledgments
We are grateful to the patients for their participation in this study.
Author Contributions
QZ and JZ conceptualized the study, reviewed the literature, and wrote the initial and final drafts. PG, YM, DZ, RW, YZ, SW, YC, and MZ worked on acquisition, interpretation, and analysis of data. All authors participated in critically revising the manuscript’s form and content. JZ critically revised the manuscript and approved the final version for publication.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by the National Science and Technology Major Projects of China (2015BAI12B04), the Program of the National Natural Science Foundation of China (81870904), the Program of Capital Medical University Basic-Clinical Cooperation (17JL44), the Program of Natural Science Foundation of Capital Medical University (PYZ2017068), and the project of study abroad of Beijing Tiantan hospital.
Ethical Approval
The Ethics Committee of Beijing Tiantan Hospital, Capital Medical University, Beijing, China, approved the study.