
Abstract
Background
POLG pathogenic variants are the commonest single-gene cause of inherited mitochondrial disease. However, the data on clinicogenetic associations in POLG-related disorders are sparse. This study maps the clinicogenetic spectrum of POLG-related disorders in the pediatric population.
Methods
Individuals were recruited across 6 centers in India. Children diagnosed between January 2015 and August 2020 with pathogenic or likely pathogenic POLG variants and age of onset <15 years were eligible. Phenotypically, patients were categorized into Alpers-Huttenlocher syndrome; myocerebrohepatopathy syndrome; myoclonic epilepsy, myopathy, and sensory ataxia; ataxia-neuropathy spectrum; Leigh disease; and autosomal dominant / recessive progressive external ophthalmoplegia.
Results
A total of 3729 genetic reports and 4256 hospital records were screened. Twenty-two patients with pathogenic variants were included. Phenotypically, patients were classifiable into Alpers-Huttenlocher syndrome (8/22; 36.4%), progressive external ophthalmoplegia (8/22; 36.4%), Leigh disease (2/22; 9.1%), ataxia-neuropathy spectrum (2/22; 9.1%), and unclassified (2/22; 9.1%). The prominent clinical manifestations included developmental delay (n = 14; 63.7%), neuroregression (n = 14; 63.7%), encephalopathy (n = 11; 50%), epilepsy (n = 11; 50%), ophthalmoplegia (n = 8; 36.4%), and liver dysfunction (n = 8; 36.4%). Forty-four pathogenic variants were identified at 13 loci, and these were clustered at exonuclease (18/44; 40.9%), linker (13/44; 29.5%), polymerase (10/44; 22.7%), and N-terminal domains (3/44; 6.8%). Genotype-phenotype analysis suggested that serious outcomes including neuroregression (odds ratio [OR] 11, 95% CI 2.5, 41), epilepsy (OR 9, 95% CI 2.4, 39), encephalopathy (OR 5.7, 95% CI 1.4, 19), and hepatic dysfunction (OR 4.6, 95% CI 21.3, 15) were associated with at least 1 variant involving linker or polymerase domain.
Conclusions
We describe the clinical subgroups and their associations with different POLG domains. These can aid in the development of follow-up and management strategies of presymptomatic individuals.
Introduction
Mitochondrial DNA (mtDNA) encodes 13 proteins essential for the electron transport chain that provides most of the ATP in the cell. The replication/biogenesis of mtDNA is orchestrated by several proteins, including DNA polymerase γ (Polγ), Twinkle mtDNA helicase, topoisomerases, and RNaseH. 1 Human Polγ is governed by 2 nuclear subunits, POLG (also known as POLG1, 140kD) and POLG2 (55 kD). POLG (OMIM*174763) on chromosome 15 encodes the mtDNA polymerase's catalytic subunit, the enzyme responsible for mtDNA repair and replication.2,3 POLG2, encoded by POLG2 on chromosome 17, enhances polymerase processivity by increasing the catalytic subunit's affinity for DNA.4–6
Polγ failure, therefore, results in mtDNA depletion and accumulation of multiple mtDNA deletions in postmitotic tissues such as the nervous system, muscles, and liver. 6 van Goetham, in 2001, first described 4 mutations in POLG associated with either autosomal dominant progressive external ophthalmoplegia (adPEO) or autosomal recessive PEO (arPEO). 7 Clinically, POLG pathogenic variants have been associated with a wide range of overlapping phenotypes, including (1) Alpers-Huttenlocher syndrome; (2) myocerebrohepatopathy syndrome; (3) myoclonic epilepsy, myopathy, and sensory ataxia; (4) ataxia-neuropathy spectrum, including previously described sensory ataxia, neuropathy, dysarthria, and ophthalmoplegia and mitochondrial recessive ataxia syndrome; (5) adPEO; and (6) arPEO.5,8 The cornerstone of clinical diagnosis is a recognizable phenotypic classification. As is evident from the details above, POLG variants are associated with overlapping phenotypes, which makes the clinical diagnosis of POLG-related disorders extremely challenging.
Exploring the POLG-related clinical disease patterns and their correlation with genotypes can help improve our understanding of the functions of POLG and aid the development of management strategies for presymptomatic and symptomatic individuals. In addition, quantifying the correlation of different POLG pathogenic variants with clinical findings can help in targeting specific genetic variants in the specific clinical scenario. Since 2001, >300 POLG-related pathogenic variants have been described (http://tools.niehs.nih.gov/polg/). However, most POLG-related data has emerged from developed countries, mainly Norwegian and Finnish, whereas data from the Indian subcontinent is scant. In this context, this multicentric study was undertaken to study the clinical and genetic findings of 22 Indian children with POLG pathogenic variants.
Materials and Methods
Patients
This study was carried out at 6 tertiary care referral centers across India. Recruitment was done via one of the following options: (1) individuals diagnosed via exome/Sanger sequencing at any of these centers; or (2) individuals who had got their exome sequencing done elsewhere but are followed by one of the coauthors. For option 1, all genetic testing done in each of these centers from January 1, 2015, to August 31, 2020, was screened, and for option 2, the hospital records from January 1, 2015, to August 31, 2020, were screened to identify patients with POLG variants. All children, recruited through option 1 or 2, with variants in POLG classified as pathogenic or likely pathogenic according to the American College of Medical Genetics and Genomics guidelines for the interpretation of sequence variants and age of onset <15 years were included. Patients with incomplete data or presymptomatic diagnosis due to family screening were excluded. Patient data, including demography, clinical features, laboratory data, neurophysiological characteristics, neuroimaging features, genetic variants, and therapeutic interventions, were retrieved from hospital medical records and chronicled using a case record form. Written informed consent was obtained from parents and caregivers. The study was performed with approval from the Institutional Ethical Committees of all participating centers.
Identification of POLG variants
POLG variants were identified using exome sequencing and confirmed by Sanger sequencing. The carrier status of parents was confirmed using Sanger sequencing for identified variants. Segregation analysis in first-degree relatives was performed in all except 1 (sibling to be tested had died). All POLG variants were annotated according to the Ensembl variant effect predictor in August 2020. 9
Phenotypic classifications
Based on clinical and laboratory features, patients were classified into 6 clinical groups. First, Alpers-Huttenlocher syndrome was defined by the clinical triad of (1) refractory seizures that often had a focal component, (2) psychomotor regression that was often episodic and triggered by intercurrent infection, and (3) hepatopathy with or without acute liver failure. In addition, characteristic liver histopathologic changes suggestive of Alpers-Huttenlocher syndrome included 2 of the following: microvesicular steatosis, bile ductular proliferation, hepatocyte dropout, bridging fibrosis or cirrhosis, the collapse of liver cell plates, parenchymal lobular architecture, regenerative nodules, and oncocytic changes in scattered hepatocytes not affected by steatosis.5,10 Second, myocerebrohepatopathy syndrome was defined by the clinical triad of (1) myopathy/hypotonia, (2) developmental delay/encephalopathy, and (3) liver dysfunction. In addition, diagnostic criteria for myocerebrohepatopathy syndrome included absence of hepatic histopathological features of classical Alpers or at least 2 of the following: (1) neuropathy; (2) seizures; (3) elevated blood or cerebrospinal fluid lactate; (4) dicarboxylic aciduria; (5) renal tubular dysfunction with aminoaciduria, glycosuria, or bicarbonaturia; (6) hearing loss; (7) abnormal magnetic resonance imaging (MRI), with either cerebral volume loss, delayed myelination, or white matter disease; and (8) either an isolated deficiency of complex IV or a combined defect of 2 or more oxidative phosphorylation complexes in skeletal muscle or liver biopsy. 5 Third, ataxia-neuropathy spectrum included an overlapping spectrum of disorders having ataxia and neuropathy in the absence of significant muscle weakness or myopathy. 6 Fourth, myoclonic epilepsy, myopathy, and sensory ataxia included the disorders presenting with myopathy, epilepsy, and ataxia in the absence of ophthalmoplegia. 6 Lastly, arPEO and adPEO included patients with ophthalmoplegia with recessive and dominant inheritance, respectively. PEO+ was used if patients had any additional findings, including sensory ataxia, neuropathy, or dysarthria. If the patient was unclassifiable into any of the above groups, he or she was assigned “Unclassifiable.”
Molecular Modeling
PyMOL (DeLano Scientific) was used to visualize and model amino acids in the 3-dimensional model of the human POLG. 11 The model was generated using PDB: 4ZTU. 12
Statistical Analysis
Summary statistics, including a median and interquartile range for continuous variables and proportions for categorical variables, were calculated for the clusters having ≥5 patients. Post hoc analysis was performed for clusters of phenotypic features within the genotypic spectrum of POLG variants. The predominant phenotypic feature was taken as input, and its association with the domain of identified pathogenic variant was determined using the Fisher exact test. The continuous variables were compared using the Mann-Whitney test. Statistical analysis was conducted using R, version 3.5.1 (R Foundation).
Data Availability
The data analyzed in this study are available from the corresponding author on request.
Results
Genetic Characteristics
A total of 3729 genetic reports were screened, and 24 patients were identified. In addition, 2 patients were included from the screening of 4256 hospital records. Overall, we identified 26 patients from 25 families with variants in POLG. Based on our genetic curation criteria, 22 patients with pathogenic or likely pathogenic variants were included, and 4 patients with variant of unknown significance were excluded. Eight of the 22 patients (36.4%) were from consanguineous families, and 10 of 22 patients (45.5%) came from families with affected family members (mother = 2, siblings = 7, cousin = 1).
We identified 44 pathogenic variants (including 1 novel variant) at 14 different loci, all of which were missense variants. The POLG gene was divided into the following domains: (a) N-terminal domain including the mitochondrial targeting sequence: amino acid 1-170; (b) exonuclease domain: amino acid 171-440; (c) linker domain: amino acid 476-785; and (d) polymerase domain: amino acid 816-1239. The spread of pathogenic variants indicated the following clustering: N-terminal domain including the mitochondrial targeting sequence (2 patients with 3 pathogenic variants [6.8%]), exonuclease domain (11 patients with 18 pathogenic variants [40.9%]), linker domain (9 patients with 13 pathogenic variants [29.5%]), and polymerase domain (9 patients with 10 pathogenic variants [22.7%]) (Figure 1). The alleles were homozygous in 16 (72.7%) and compound heterozygous in 6 (27.3%) patients suggesting autosomal recessive transmission in the cohort.
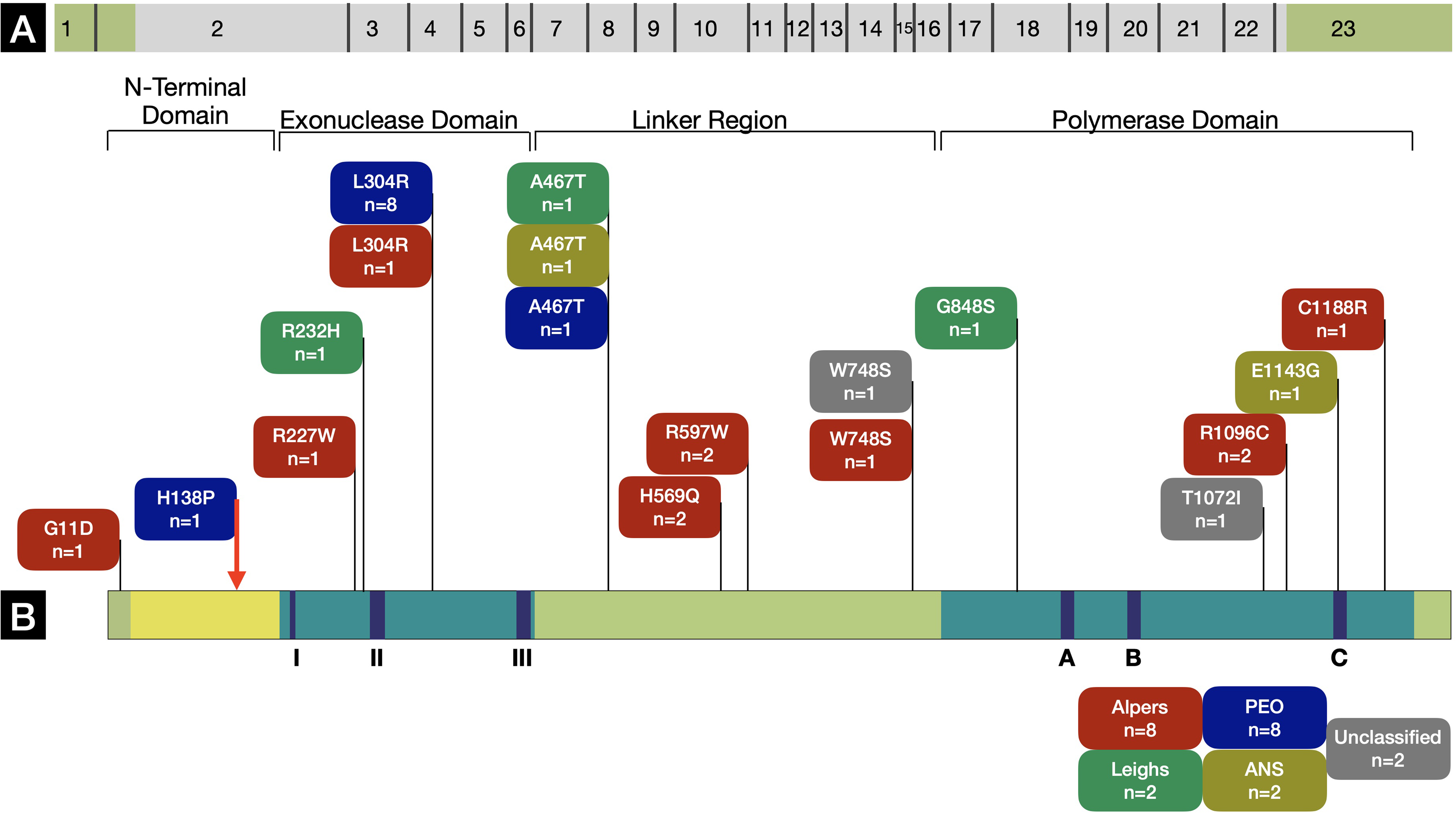
(A and B) Schematic diagram of the POLG gene illustrating pathogenic variants identified in this study. The novel pathogenic variant has been highlighted with a red arrow. The representation has been adapted from an illustration at http://tools.niehs.nih.gov/polg/. The adaptation has been published with permission from Dr Bill Copeland. (AHS, Alpers-Huttenlocher syndrome; PEO, progressive external ophthalmoplegia)
One novel missense variant (c.413T>C) was identified with H138P amino acid change. It is an invariant residue conserved from yeast to human. The histidine residue was found near the end of an alpha-helix near the exonuclease domain and analysis of protein structure suggested that a change to proline would cause a kink in the helix and alter the local structure. This variant, like others in this area, would likely be pathogenic but recessive. Our patient (Supplementary Table 1, patient 2, phenotypically PEO) was compound heterozygote for this variant along with c.911T>G (L304R) variant. Segregation analysis revealed that mother was an asymptomatic heterozygote for H138P and father was an asymptomatic heterozygote for L304R. The 3D structure of POLG with amino acids of interest highlighted has been illustrated in Figure 2A. The novel histidine 138 has been illustrated in Figure 2C and D. The predicted effect of each identified variant on POLG structure and function has been summarized in Figure 2B.6,13–18
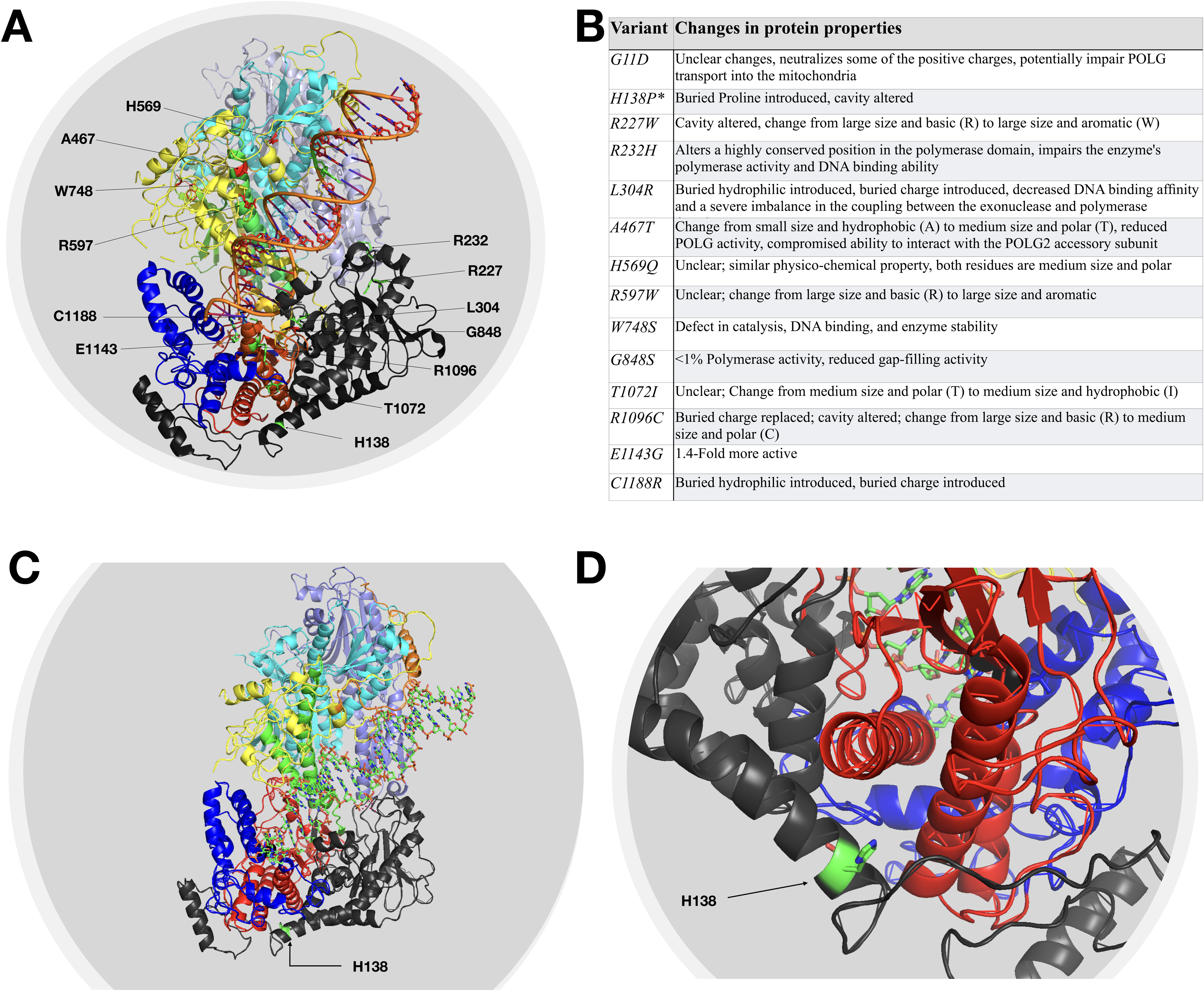
The figure depicts the molecular modeling of the human DNA polymerase gamma. The amino acids of interest have been highlighted. Panel A highlights the amino acids that have been identified to be substituted in the study. The table in panel B illustrates the impact of identified variants on protein characteristics. The novel variant H138P has been marked with an asterisk. Panels C and D display the rotated and close-up view illustrating the H138 locus.
Phenotypic Characteristics
Clinically the patients were classified into Alpers-Huttenlocher syndrome (n = 8; 36.4%), PEO/PEO+ (n = 8; 36.4%), Leigh disease (n = 2; 9.1%), ataxia-neuropathy spectrum (n = 2; 9.1%), and unclassified (n = 2; 9.1%) based on definitions mentioned above. None of the patients fulfilled the defining criteria for myocerebrohepatopathy syndrome or myoclonic epilepsy, myopathy, and sensory ataxia. Summary statistics were calculated only for patients with Alpers-Huttenlocher syndrome and progressive external ophthalmoplegia. The prominent neurologic manifestations included developmental delay (n = 14; 63.7%), neuroregression (n = 14; 63.7%), encephalopathy (n = 11; 50%), epilepsy (n = 11; 50%), hypotonia (n = 10; 45.5%), dystonia (n = 3; 13.6%), strokelike episodes (n = 1; 4.5%), and hearing impairment (n = 1; 4.5%). Other systemic manifestations included ophthalmoplegia (n = 8; 36.4%), optic atrophy (n = 3; 13.6%), nystagmus (n = 2; 9.1%), liver dysfunction (n = 8; 36.4%), faltering growth (n = 16; 72.7%), vomiting (n = 12; 54.5%), chronic diarrhea (n = 2; 9.1%), renal tubular dysfunction (n = 2; 9.1%), and hypoglycemia (n = 3; 13.6%). The median (interquartile range) age for onset of Alpers syndrome (8.5 [4-36] months) was significantly less as compared to that for progressive external ophthalmoplegia (53 [39-68] months; P = .002). The clinical details of patients have been summarized in Table 1 and details pertaining to individual patients are highlighted in supplementary Table 1.
Clinical Features of the Study Group. a
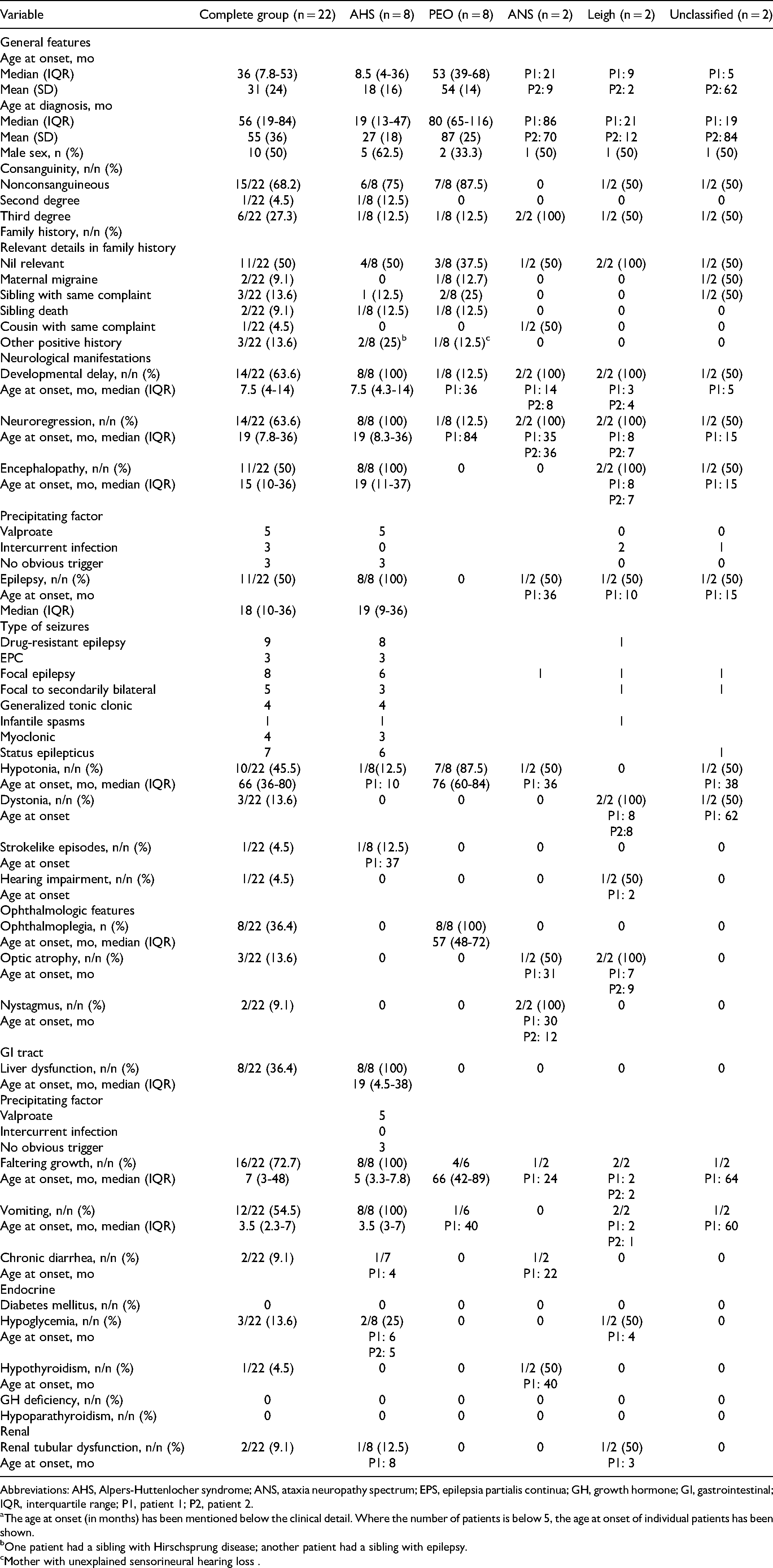
Abbreviations: AHS, Alpers-Huttenlocher syndrome; ANS, ataxia neuropathy spectrum; EPS, epilepsia partialis continua; GH, growth hormone; GI, gastrointestinal; IQR, interquartile range; P1, patient 1; P2, patient 2.
The age at onset (in months) has been mentioned below the clinical detail. Where the number of patients is below 5, the age at onset of individual patients has been shown.
One patient had a sibling with Hirschsprung disease; another patient had a sibling with epilepsy.
Mother with unexplained sensorineural hearing loss .
Investigations
Brain MRI was performed for 20/22 patients. Brain MRI was normal in all patients with progressive external ophthalmoplegia. Generalized cortical atrophy was noted in 4 of 20 patients (20%), whereas 6 of 20 patients (30%) demonstrated focal cortical lesions. Basal ganglia, thalamic, and brain stem lesions were observed in 5 (25%), 3 (15%), and 4 patients (20%), respectively. Elevated lactate was demonstrated in 6 of 16 patients (37.5%) in blood and 4 of 11 patients (36.4%) in cerebrospinal fluid. Hepatic aminotransferases were increased in 9 of 19 cases (47.4%). Hypoalbuminemia and hyperCKemia were noted in 8 of 19 (42.1%) and 5 of 18 patients (27.8%), respectively. EEG was performed in 17 patients, of which 11 (64.7%) showed epileptiform activity. The details pertaining to investigations have been summarized in Table 2, and investigations in individual patients are illustrated in Supplementary Table 2.
Details of Laboratory, Electroencephalographic and Neuroimaging Characteristics of Study Group.
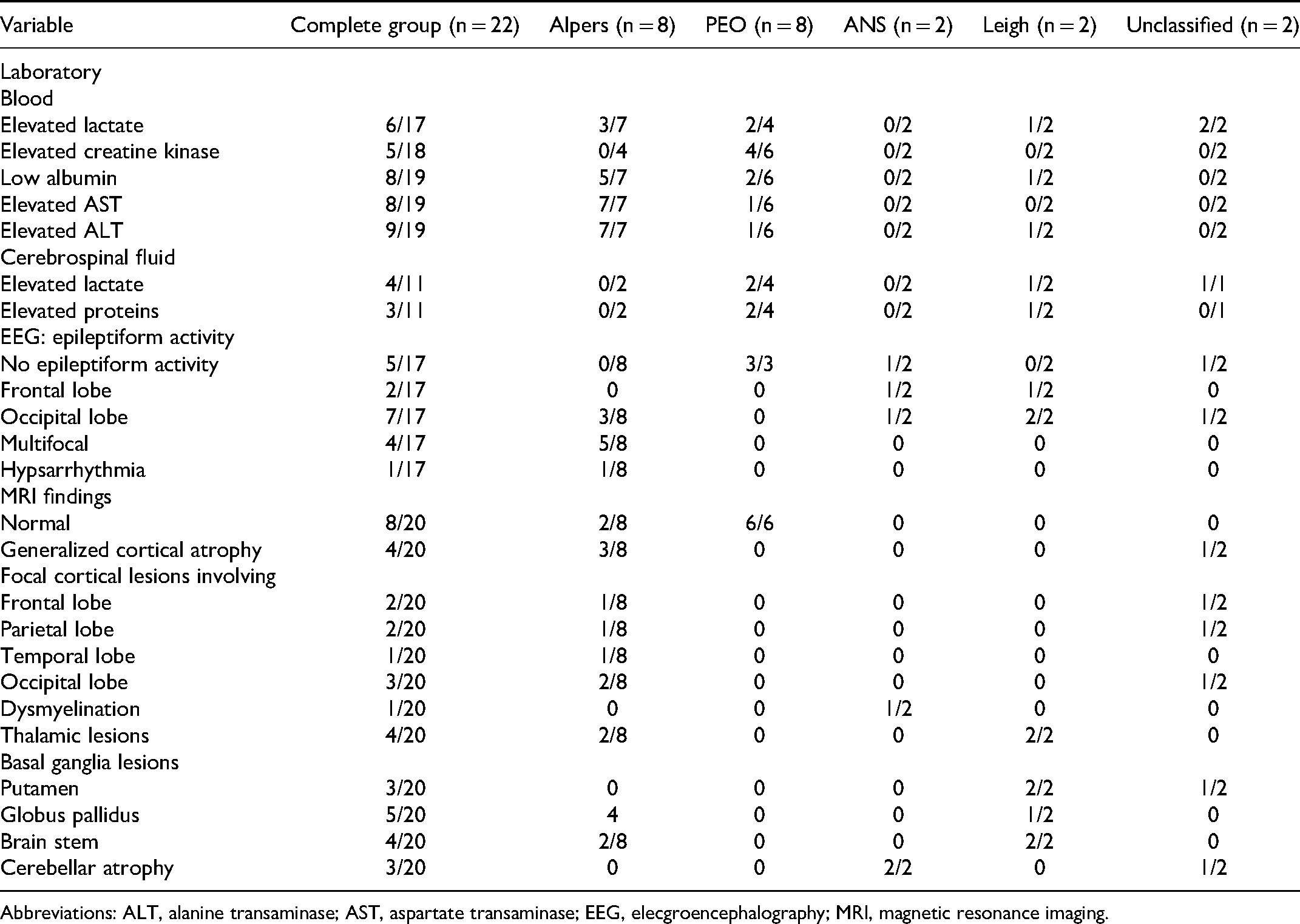
Abbreviations: ALT, alanine transaminase; AST, aspartate transaminase; EEG, elecgroencephalography; MRI, magnetic resonance imaging.
Genotype-Phenotype Correlation
A cluster analysis was performed using the predominant phenotypic features to identify the possible phenotypic clusters within the genetic pleiotropy of POLG pathogenic variants. Ophthalmoplegia, myopathy, developmental delay, neuroregression, epilepsy, encephalopathy, dystonia, ataxia, and neuropathy were used as input variables for association with the genetic domain where the pathogenic variants are clustered (N-terminal, exonuclease, linker, and polymerase). In this exploratory analysis, we noted that ophthalmoplegia (odds ratio [OR] 42, 95% CI 6.4, 209; P = .003) and myopathy (OR 24, 95% CI 4.1, 118; P = .003) were significantly associated with pathogenic variants involving the exonuclease domain. Developmental delay (OR 11, 95% CI 1.4, 127; P = .03) and neuroregression (OR 11, 95% CI 1.4, 127; P = .03) were significantly associated with pathogenic variants in the linker domain. Further grouped analysis suggested that serious outcomes including developmental delay (OR 11, 95% CI 2.5, 41; P = .001), neuroregression (OR 11, 95% CI 2.5, 41; P = .001), epilepsy (OR 9, 95% CI 2.4, 39; P = .002), encephalopathy (OR 5.7, 95% CI 1.4, 19; P = .015), and hepatic dysfunction (OR 4.6, 95% CI 21.3, 15; P = .03) were significantly associated with at least 1 pathogenic variant involving linker or polymerase domain (Table 3). There was no consistent association between the other phenotypic features and genetic variants.
Clinicogenetic Associations With Target Motifs Among Identified Mutations (Total ng = 44; Total np = 22). a
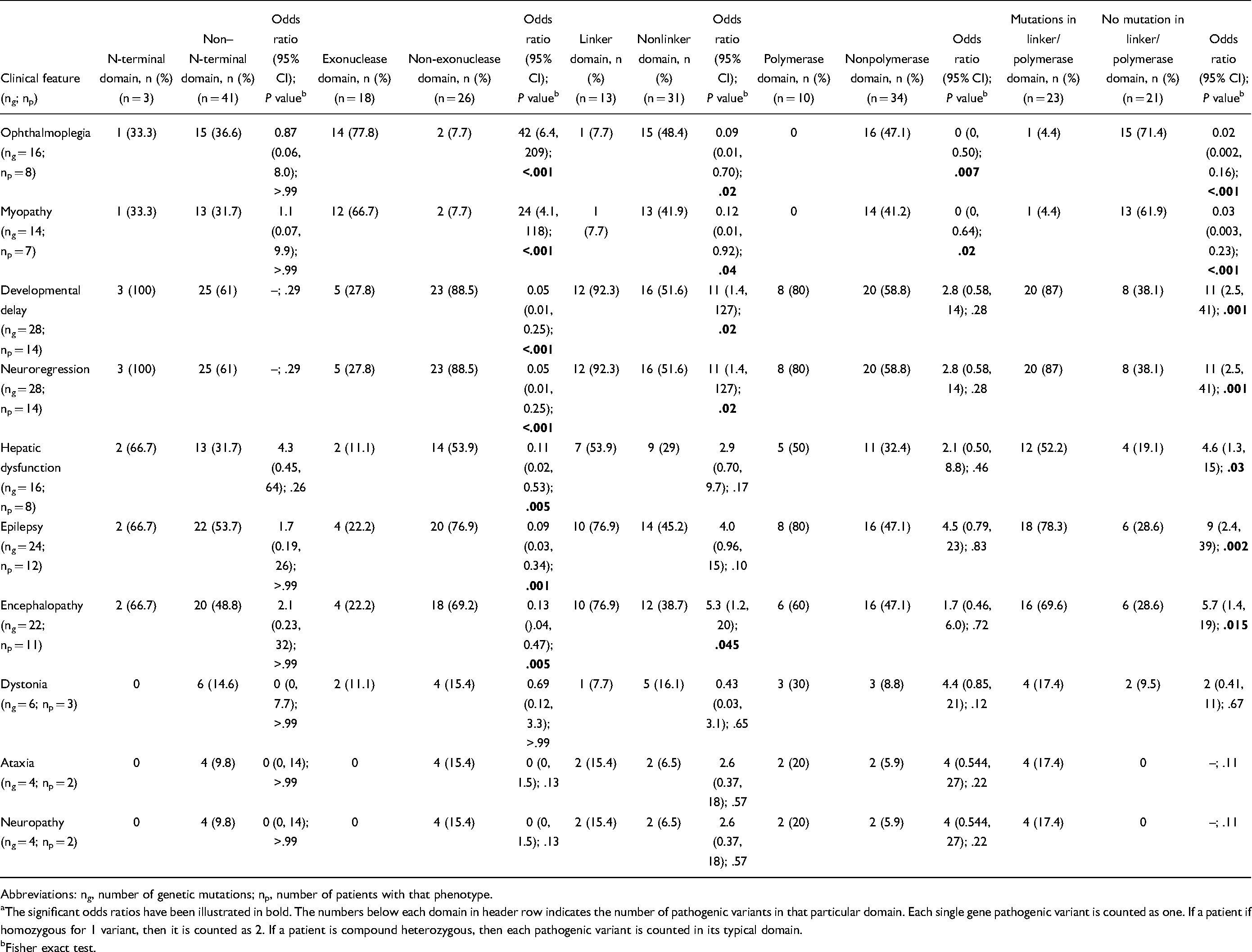
Abbreviations: ng, number of genetic mutations; np, number of patients with that phenotype.
The significant odds ratios have been illustrated in bold. The numbers below each domain in header row indicates the number of pathogenic variants in that particular domain. Each single gene pathogenic variant is counted as one. If a patient if homozygous for 1 variant, then it is counted as 2. If a patient is compound heterozygous, then each pathogenic variant is counted in its typical domain.
Fisher exact test.
Discussion
This multicentric study presents phenotype and genotype data among 22 children with POLG pathogenic variants. Genotype-phenotype correlations are explored based on variant localization. Our study, the first reporting POLG mutational landscape from Indian subcontinent and among the few reporting POLG related clinicogenetic correlations, characterizes POLG-related disease as a multisystem disorder with predominantly neurologic (encephalopathy, epilepsy, neuroregression), neuromuscular (ptosis, myopathy), and/or hepatic dysfunction. 19 Phenotypically, our cohort had 2 minor groups of Alpers-Huttenlocher syndrome and progressive external ophthalmoplegia with 8 patients each. Alpers-Huttenlocher syndrome typically had an earlier onset, with nearly 75% of children presenting below 3 years with encephalopathy, hepatic dysfunction, or epilepsy. All the patients with Alpers-Huttenlocher syndrome developed encephalopathy (median age = 19 months), and in 5 of 8 patients, encephalopathy was precipitated by valproate administration. In addition, all patients with Alpers-Huttenlocher syndrome manifested epilepsy (median age = 19 months). The most common seizure semiology included focal seizure, and in 3 patients epilepsia partialis continua was also present. Developmental delay and neuroregression were present in all patients with Alpers-Huttenlocher syndrome. In contrast, progressive external ophthalmoplegia was characterized by a milder spectrum of disease, with the predominant clinical feature being ophthalmoplegia. The median age of progressive external ophthalmoplegia onset was 50 months, which was significantly more than that for Alpers-Huttenlocher syndrome. The other 3 smaller clusters of 2 patients each included Leigh syndrome, ataxia-neuropathy spectrum, and unclassified spectrum who manifested with features of encephalopathy, epilepsy, dystonia, and ataxia. Studies have demonstrated that clinical manifestations of POLG pathogenic variants can be heterogenous in terms of age of presentation (neonatal to adulthood) and type of presentation (severe manifestations like encephalopathy, epilepsy, hepatic failure to relatively mild presentations including ophthalmoplegia, ataxia, and myopathy).2,5,13,20 The clinical spectrum observed in our cohort is similar to earlier reports suggesting that the POLG phenotypic spectrum consists of early-onset severe form, later-onset milder form, and multiple other variable expressions of variable severity and diverse symptomatology. 2
Genetically, our study cohort confirms that pathogenic variants are scattered throughout the coding region (Figure 1).2,5 Conventionally POLG gene is divided into 5 distinct domains implying a genotype-phenotype relationship at the biochemical level. However, the same cannot be extrapolated to the clinical phenotype.2,15 We demonstrate 44 pathogenic variants distributed between N-terminal domain (n = 3), exonuclease domain (n = 18), linker domain (n = 13), and polymerase domain (n = 10) among 22 patients. The extrapolatory analyses indicated that ophthalmoplegia and myopathy were more likely if the patient had at least 1 pathogenic variant in the exonuclease domain. Also, neuroregression was more likely if the patient had at least 1 variant in the linker domain. Further analysis suggested that most severe diseases (epilepsy, encephalopathy, neuroregression, developmental delay, and hepatic dysfunction) were associated with at least 1 variant in the linker domain or the polymerase domain. However, beyond these preliminary associations, we could not demonstrate any specific genotype-phenotype association. POLG pathogenic variants in different domains may have a differential effect on enzymatic stability and the catalytic efficiency of POLG, and hence may be responsible for pleiotropy exhibited with POLG. Most earlier reports have looked at phenotypic correlations with individual pathogenic variants and not with the domain harboring the variant. In a study describing the clinical disease associated with A467T and W748S in 26 patients, the authors reported a spectrum varying from early-onset Alpers-Huttenlocher syndrome to late-onset ataxia. 20 Our cohort had 3 patients with A467T variant (1 each with progressive external ophthalmoplegia, ataxia-neuropathy spectrum, and Leigh disease) and 2 patients with W748S variant (Alpers-Huttenlocher syndrome = 1; unclassifiable = 1). Most of the other reports have failed to establish a clear phenotypic-genotypic association.5,13 Our findings, in concordance with earlier reports, imply that all children with unexplained encephalopathy or neurologic disorders with multisystem involvement be screened for pathogenic variants involving POLG gene.2,21
Our study was limited by its retrospective design, lack of phenotype-matched controls, and small cohort size. However, the reports detailing clinical and molecular characteristics of POLG-related disorders are scarce. Despite limitations, this study demonstrates a novel H138P variant near the exonuclease domain. The variant in this conserved residue is likely to be pathogenic. In addition, we have attempted to determine the genotype-phenotype correlations in this rare group of disorder. Our findings suggest that though POLG-related disorders’ phenotype is complex and overlapping, they predominantly include the more severe early-onset variety and a milder phenotype that has its onset in late childhood. The severe phenotype is more likely to be associated with a variant involving the linker or polymerase domain. Besides, our findings highlight that in children with unexplained encephalopathy, epilepsy, hepatic dysfunction, or multisystem disorder, either in isolation or in combination, the POLG gene should be sequenced.
Supplemental Material
sj-docx-1-jcn-10.1177_08830738211067065 - Supplemental material for Clinical and molecular spectrum associated with Polymerase-γ related disorders
Supplemental material, sj-docx-1-jcn-10.1177_08830738211067065 for Clinical and molecular spectrum associated with Polymerase-γ related disorders by Ruchika Jha, Harshkumar Patel, Rachana Dubey, Jyotindra N. Goswami, Chandana Bhagwat, Lokesh Saini, Ranjith K. Manokaran, Biju M. John, Uday B. Kovilapu, Aneesh Mohimen, Apoorv Saxena and Vishal Sondhi in Journal of Child Neurology
Supplemental Material
sj-docx-2-jcn-10.1177_08830738211067065 - Supplemental material for Clinical and molecular spectrum associated with Polymerase-γ related disorders
Supplemental material, sj-docx-2-jcn-10.1177_08830738211067065 for Clinical and molecular spectrum associated with Polymerase-γ related disorders by Ruchika Jha, Harshkumar Patel, Rachana Dubey, Jyotindra N. Goswami, Chandana Bhagwat, Lokesh Saini, Ranjith K. Manokaran, Biju M. John, Uday B. Kovilapu, Aneesh Mohimen, Apoorv Saxena and Vishal Sondhi in Journal of Child Neurology
Footnotes
Acknowledgments
We thank the patients and relatives for their participation in this study. We are grateful to Dr Bill Copeland for his assistance with developing the 3D protein structure for POLG and illustrating the effect of novel variant (H138P). We thank Dr Naveen Sankhyan, Department of Pediatrics, Pediatric Neurology Unit, Postgraduate Institute of Medical Education and Research, Chandigarh, India, for his inputs related to the manuscript.
Authors Contributions
RJ and HP were equally responsible for the work described in this article. RJ, HP, LS, RD, JNG, and VS conceived and designed the study. RJ, HP, RD, JNG, CB, LS, RKM, BMJ, UBK, AM, AS, and VS contributed to the acquisition and analysis of data. RJ, HP, LS, and VS drafted the text and prepared the figures.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship and/or publication of this article.
Ethical Approval
The study was performed with approval from the Institutional Ethical Committees of all participating centers individually and was approved centrally by Institutional Ethical Commitee of Armed Forces Medical College, Pune (IEC Serial Number IEC/2020/73).
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.