
Abstract
The aim of this study was to describe the phenotype of Leber hereditary optic neuropathy occurring in pediatric females. This disease generally affects young adult males, but it can occur also in females, and research data in this population is lacking. The very early onset can challenge the diagnosis and delay treatment. We searched PubMed through February 2021 and identified 226 pediatric females with genetically confirmed Leber hereditary optic neuropathy and added a new case of a 3-year-old female. The male-female ratio was 1.8:1; the mean onset age in females was 11 years with the onset at 3 years of age occurring in 3 females only. Acute onset with mild visual impairment was the most common presentation, associated with optic disc edema in 16%. Differential diagnoses are pseudotumor cerebri, optic nerve drusen and optic neuritis. The outcome is poor with partial recovery in 50%, despite some receiving Idebenone therapy.
Leber hereditary optic neuropathy (LHON) is a mitochondrial genetic disease that leads to painless, acute or subacute loss of vision over a period of weeks to months. Affected individuals usually carry one of 3 more common disease-causing variants in the mitochondrial genes ND1 m.3460G>A, ND4 m.11778G>A, ND6 m.14484T>C, but more than 30 mitochondrial DNA genetic variants play a role in disease expression. 1 The penetrance of these genetic defects is incomplete and is influenced by the phenomenon of heteroplasmy, mitotic segregation, threshold effect, by effect of other modulatory genes and by epigenetic factors, making genetic counseling extremely difficult. 1
Leber hereditary optic neuropathy occurs in approximately 1:30 000 people in the general population. 2 Males are mostly affected in their second or third decade of life. Patients with classic Leber hereditary optic neuropathy usually experience visual loss between 15 and 35 years of age, and childhood onset occurs in 11.5% of cases. 3 In adults, the female/male ratio is 1:5. 4 The median age for childhood onset is variable according to different studies: the mean age at onset of the disease is approximately 6.8 years (range 2-11 years) in European centers and approximately 10 years (range 3.5-17) in the Chinese population.2,5,6
In childhood, males are also more affected than females.2,5,6 The mean age of onset in pediatric females is not described in literature.
The clinical presentation of pediatric Leber hereditary optic neuropathy is distinguishable in 3 forms: acute, slowly progressive, and subclinical. 5 Information on the phenotype in female children is scarce, particularly when onset is at a very young age. When ultrarare phenotypes are involved, the diagnosis is challenged by other similar and more frequent pediatric neurologic and neuro-ophthalmologic conditions, such as optic nerve drusen, optic neuritis, and other optic neuropathies.7,8 Treatment with Idebenone has been shown to counteract the progression of the disease toward blindness in many patients, but it has to be started early in the disease course.9–11 Idebenone is a strong antioxidant and inhibitor of lipid peroxidation that interacts with the electron transport chain, facilitating mitochondrial electron flux and bypassing complex I.10,11
The aim of this study was to describe the phenotype of Leber hereditary optic neuropathy occurring in pediatric females by identifying the cases reported in the literature. The focus was on very early onset, and we added a new case with onset at 3 years of age.
Materials and Methods
We conducted a literature search for pediatric cases of Leber hereditary optic neuropathy occurring in females.
We searched case series, case reports, and retrospective and prospective English-language studies reported in PubMed (PubMed and PubMed Central) through February 2021. The search term combinations were “Childhood Leber Hereditary Optic Neuropathy AND females,” “Leber mitochondrial Optic Neuropathy in pediatrics AND females,” and “Leber Hereditary Optic Neuropathy AND girls,” the last to consider adult female cases with pediatric onset. Our search algorithm is illustrated in Figure 1. All the articles were screened by reading titles, abstracts, and, as needed, text.
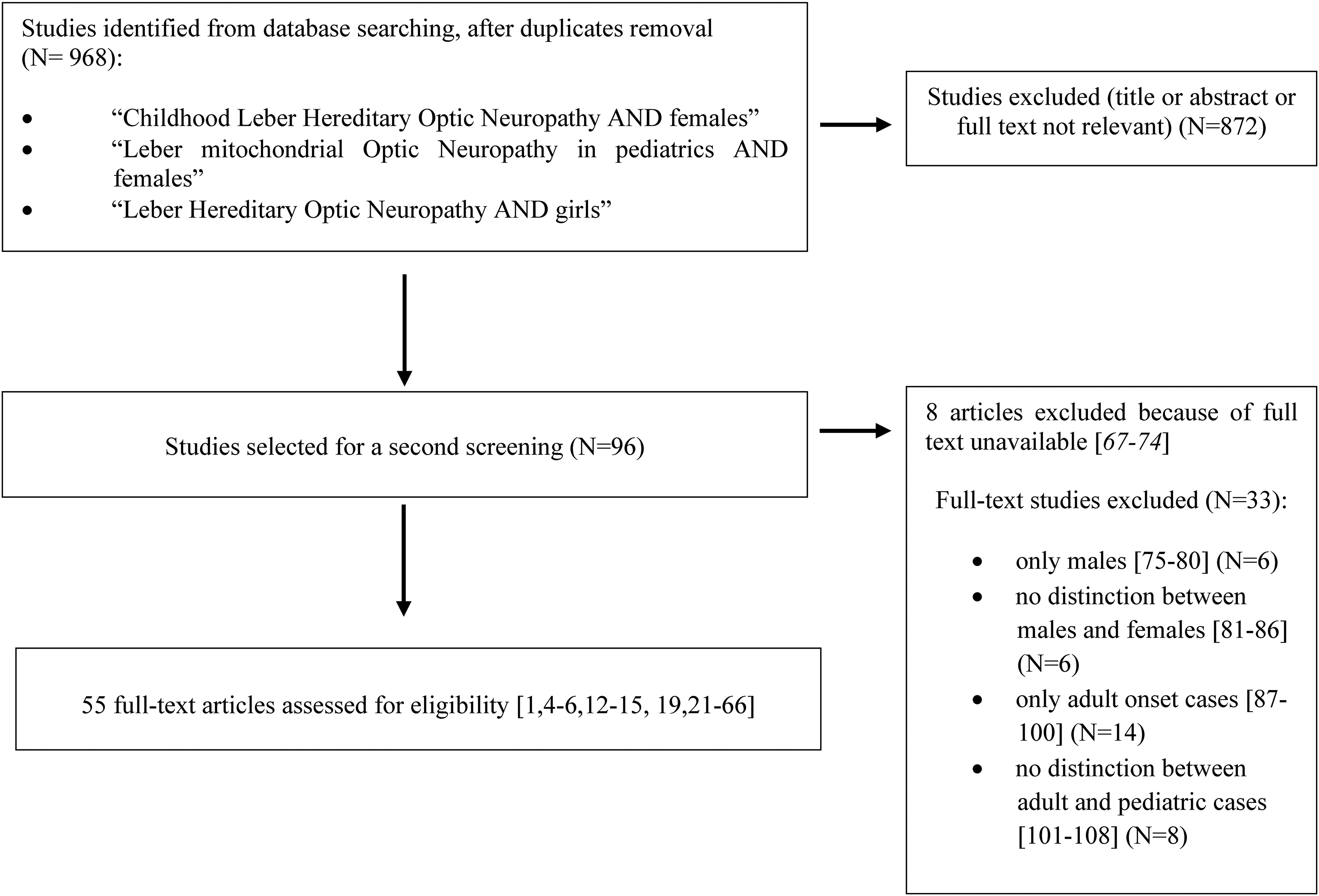
Literature search algorithm: article exclusion process that resulted in the selection of 55 articles, as included in our database.
All studies reporting cases of Leber hereditary optic neuropathy with onset during pediatric age (0-18 years) were considered for inclusion. The onset was defined by the first ophthalmologic examination confirming visual acuity loss.
After duplicate removal, 968 articles were screened. Fifty-five articles were eligible and analyzed to extract relevant clinical information.1,4–6,12–60
All articles with no mention of clinical cases (n = 872) were excluded. The full text remained unavailable in eight of them, so we extracted available information from the abstract.61–68 Among articles describing cases (n = 96), those regarding only male children (for a total of 8 males) were excluded.69–74 We also excluded articles including large series of patients not reporting the difference between males and females,75–80 cases with only adult onset,81–94 and those with lack of information about age at onset.95–102
For every report included, we retrieved the type of study, number of pediatric cases, and number of female cases; for female children, we extracted age of onset, type of onset, neuro-ophthalmologic data (visual acuity, fundus oculi, visual field tests, color vision, optical coherence tomography, and visual evoked potentials) and information about recovery. When measured, visual acuity was expressed in decimals. The degree of visual impairment was defined according to visual acuity as follows: mild (0.3-0.1), moderate (0.1-0.05), severe (0.05-0.02), profound (<0.02, counting fingers or light perception).36–40
Among selected cases, we identified 2 articles describing a minimum age of 3 years (very early onset).21,28
Moreover, we added to the literature data a female case from our institution with genetically confirmed Leber hereditary optic neuropathy who presented with visual impairment at 3 years of age.
Results
We identified 55 articles investigating Leber hereditary optic neuropathy in pediatric females; these individuals are described in the Supplementary Table.1,4–6,12–60
Among the included studies ,1,4–6,12–60 16 were case reports, 32 were retrospective studies, and 7 were prospective studies. In total, we included 627 Leber hereditary optic neuropathy patients with onset before 18 years, including our case. Of these, 227 were female and 400 male children, with a male-to-female ratio of 1.8:1. The mean age at disease onset of pediatric females was 11 years, with a range of 3-18 years. Only 10 of 227 female children (4.4%) had symptom onset at preschool age.13,21,28,44–46,52,60
The minimum age of onset was 3 years, described in only 3 patients, including our case (1.3%); all other females were older at onset of the visual symptoms.21,28
The onset modality of Leber hereditary optic neuropathy was described in 92 of 227 female children (40.5%): acute onset occurred in 80 of 92 (87%),1,4,5,14–16,18,20,22–24,26–37,39,41–43,46–49,53,55,58,59 subclinical or subacute onset in 4 of 92 (4%),13,19 and slowly progressive onset in 8 of 92 (9%).4–6,13,19,20
The severity of visual impairment was reported at diagnosis in 132 female children (58%). In the total population, visual impairment was bilateral with sequential involvement of the right and left eyes. The visual acuity was normal in 23 right eyes (17%) and 23 left eyes (17%).4,5,13,16,21,28,30,35,39,46,50,54,58 The visual impairment was mild in 65 right eyes (50%) and 60 left eyes (46%),1,4,5,13,15,16,20–22,24,27,28,30–32,34–37,39,42,43,45,46,48–50,52,53,56,58 moderate in 16 right eyes (12%) and 17 left eyes (13%),15,18,26,27,29–31,33,35,39,42,45,48,50,52,56 severe in 9 right eyes (7%) and 12 left eyes (9%),4,27,29,35,41,43,48,49,58 and profound in 18 right eyes (14%) and 19 left eyes (15%).4,24,27,36,39,41,45,47,49,52,57,59
Goldmann perimeter was reported in only 14 of 227 female children (6%).14,15,20,22–26,30,31,33–36 The main finding was central scotomas. Fundus oculi was reported in 37 of 227 pediatric females (16%)5,14–16,19,20,22,24–28,30,31,33,34,36,41,49: atrophy of the entire optic disc in 6 of 37,5,14,15,25,33,34 edema and hyperemia of the optic disc in 6 of 37 (patients with acute onset),20,24,26,41 atrophy of temporal sector in 1 of 37 (patient with slowly progressive onset), 5 pale optic disc in 9 of 37,16,20,22,27,28,36,49 and isolated microangiopathy in 5 of 37.19,20,30,31,41 Edema and tortuous vessels in the acute stage of Leber hereditary optic neuropathy and optic pallor or atrophy in slowly progressive Leber hereditary optic neuropathy were seen in 11 of 37 patients.5,6,14–16,19,20,22–28,30,31,33,34,36,41,49
Optical coherence tomography was described in 15 of 227 female children (6%).5,6,49,58 Those with acute onset showed diffuse thinning of the retinal nerve fiber layer, whereas those with slowly progressive onset showed atrophy of the temporal sector; in 1 case the optical coherence tomography was normal. Visual evoked potentials were reported in 19 of 227 (8%) of the female children and were characterized at disease onset by reduced amplitude and delayed latency.5,6,49,58
Data about visual recovery were reported in 27 articles and in our case, accounting for 61 of 227 pediatric females (27%).5,13,16,18–20,23,25,26,28,32,35,41–43,46,47,50,55,57–59 Seventeen of 61 female children (28%) received a specific therapy: 6 were treated with Idebenone,20,22,41,58 10 with gene therapy,35,50,54,55,57 and 1 with steroids. 26 Among those treated with Idebenone, 3 of 6 had no visual recovery,20,22 and 3 of 6 experienced stabilization or improvement of the visual defect.41,58 Ten patients underwent gene therapy: 7 of 10 (70%)35,55,57 recovered and 3 of 10 (30%) had no visual recovery.50,57 One female patient received steroids without improvement at follow-up performed 8 years later. 26 Forty-four of 61 (72%) did not receive treatment: 23 of 44 had no recovery,4,5,13,15,16,18–20,25,34,35,46 whereas 21 of 44 showed spontaneous partial recovery of visual function.5,13,16,18,19,23,28,35,43,46,47,59 Partial visual recovery, with or without therapy, globally occurred in 30 of 61 (49%) female children.5,13,16,18,19,23,28,35,41,43,46,47,55,57,59
The case added to the literature data is a pediatric female presenting with visual impairment at 3 years of age.
The parents reported normal previous psychomotor development, growth, and visual behavior. Previous personal history was not relevant, including pharmacologic therapies. Family history was negative for visual disturbances and neurologic disorders. In the previous months, the parents noticed that the girl brought her eyes close to the table during drawing, and she turned her head to favor vision with her left eye; she did not follow images on a screen, and when the right eye was covered, she complained of not seeing anything. She never complained of headache or vomiting. On arrival, she had poor visual acuity (right eye visual acuity less than 0.1 and left eye visual acuity of 0.4 on the decimal scale), and the neurologic examination was normal with the exception of vision loss. Cognitive function was normal, with the exception of visual attention skill defects. The fundus oculi examination revealed a raised optic disc with blurred edges, more pronounced in the right eye, and tortuous vessels, consistent with bilateral moderate optic disc edema. Brain magnetic resonance imaging (MRI) was normal except for mild bilateral flattening of the posterior sclera associated with mild perineural cerebrospinal fluid in the optic nerve sheath. Ultrasonographs of the posterior eye and optic nerve excluded optic nerve drusen. Given the suspicion of pseudotumour cerebri, lumbar puncture was performed and revealed an opening pressure of 200 mm H2O (normal range 100-200 mm H2O), with normal spinal fluid parameters. Blood examinations, serologic investigations, and angiotensin-converting enzyme levels were normal. Acetazolamide therapy was started with transient improvement of optic disc swelling. In the following days, the visual acuity worsened (right eye visual acuity equal to 0.1 and left eye visual acuity equal to 0.16 on the decimal scale), and eccentric fixation at distance was noted. Visual evoked potentials were performed and showed bilaterally decreased amplitudes and delayed latencies of the P100 cortical response. Cerebrospinal fluid oligoclonal bands and serum antibody testing for aquaporin-4 and myelin oligodendrocyte glycoprotein were all negative. In the hypothesis of optic neuritis, 30 mg/kg bolus of methylprednisolone was administered for 3 days, followed by a tapering course of orally administered corticosteroids. The visual evoked potentials worsened sequentially from the right to the left eye, despite steroid treatment. A second brain and orbital MRI performed 2 months later was normal, except for symmetrical signal changes in the substantia nigra of the midbrain. Considering the clinical and electrophysiological deterioration during steroid treatment, atypical for optic neuritis, despite the female sex and young age, we hypothesized Leber hereditary optic neuropathy. Molecular analysis of mitochondrial DNA revealed homoplasmy for the m.3460G>A variant in the MT-ND1 gene (R&I Genetics, Padua, Italy), suggestive of Leber hereditary optic neuropathy. The mother and sister were carriers of the same variant, in the absence of clinical symptoms. Exome analysis targeting the optic neuropathy phenotype for ruling out the co-occurrence of other forms of optic neuropathy was negative. Optical coherence tomography showed a marked reduction in the thickness of the retinal nerve fiber layer in all areas (Figure 2). Four months after disease onset, off-label Idebenone therapy was started after obtaining written informed consent by the parents to reduce mitochondrial dysfunction. The brain MRI 1 year after the first one was completely normal.
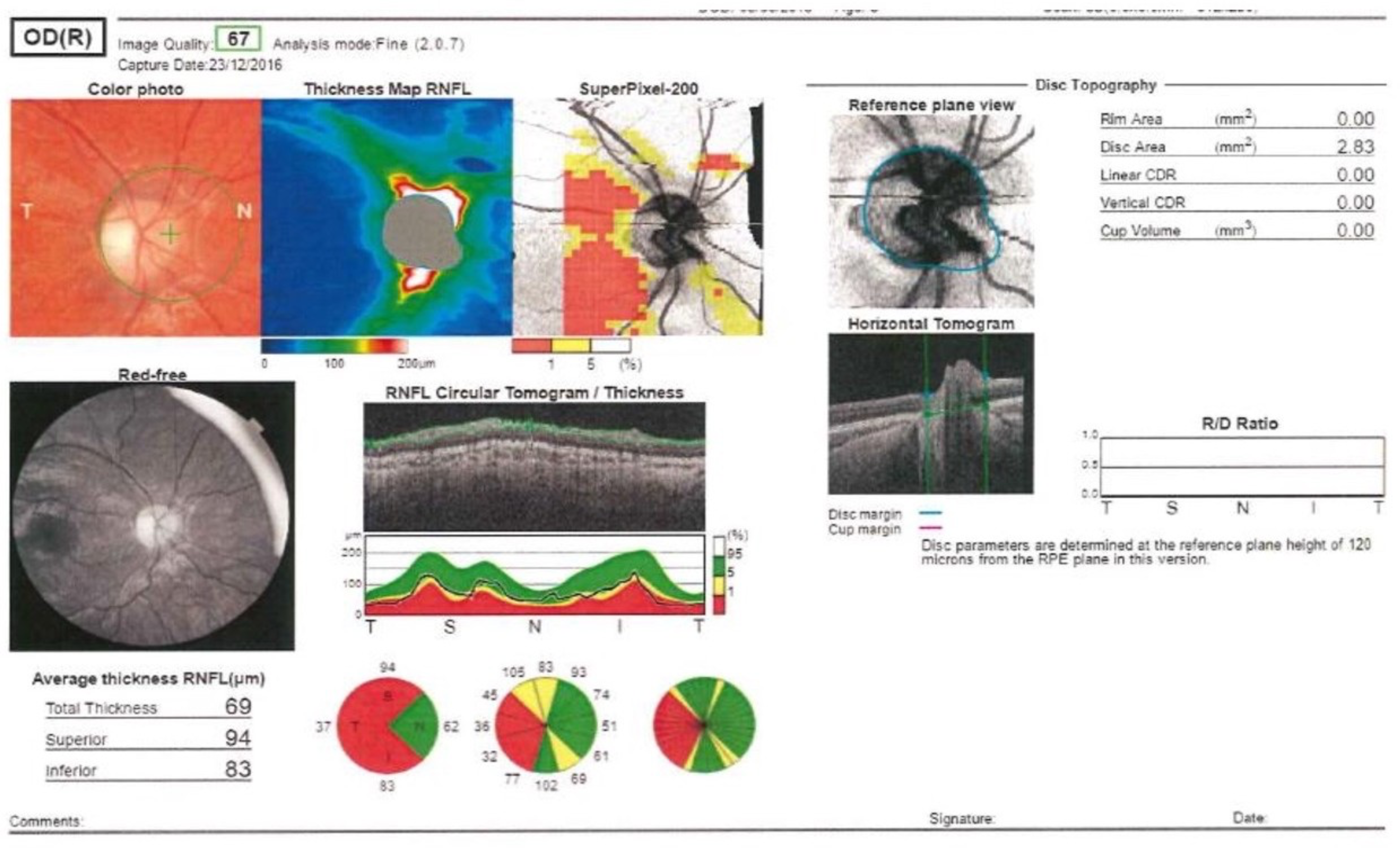
Optical coherence tomography at onset of Leber hereditary optic neuropathy found a reduction in the thickness of the retinal nerve fiber layer in all areas.
At follow-up 3 years later, during Idebenone therapy, the visual acuity improved and was left eye of 0.2 and right eye of 0.7 on the decimal scale. Color vision, tested with Ishihara tables, showed an incomplete red-green color deficiency, whereas contrast sensitivity remained abnormal. Visual field testing confirmed the presence of a central scotoma. Visual fatigue, crowding effects, and glare were unchanged, and ophthalmic symptoms hampered daily activities. Visual evoked potentials were stable over time, and optical coherence tomography showed a small progression of retinal ganglion cell atrophy (Figure 3).

Optical coherence tomography at 3-year follow-up, showing a progression of retinal ganglion cell atrophy.
Visual rehabilitation, in cooperation with the family and teachers, supported the child in the development of compensatory strategies and multisensory integration. The patient developed a compensatory posture with the head tilted on the right shoulder, right eye exotropia and hyperfunction of the left ocular muscles.
Discussion
This study reports the literature data of female children with genetically confirmed Leber hereditary optic neuropathy and adds the third case of a pediatric female presenting with abnormal visual behavior and optic disc edema at 3 years of age. We determined the male-to-female ratio, age of onset, and neuro-ophthalmologic phenotype of pediatric females, and the diagnostic challenges when the onset is very early.
Research on pediatric Leber hereditary optic neuropathy is scarce, particularly in females, and description of the phenotype is lacking, particularly at symptom onset.
In adults, the female-to-male ratio is 1:5.2,4 The male prevalence can be explained by a modulatory effect on mitochondrial expression due to some loci on the X chromosome (DXS8090-DXS1068, DXS1068-DXS8016), lifestyle (smoking exposure), and hormonal differences.7,103 In contrast, in pediatrics, this sex difference is less evident. In our study, the female-male ratio was 1:1.8. Diversity between Leber hereditary optic neuropathy phenotypes in pediatric and adult age is explainable by heavier genetic determination, nuclear modifiers, and lower hormonal and environmental influences.2,4,103,104
Childhood onset is itself a rare condition and it is poorly described in the literature. Barboni et al, in a cohort of 180 European Leber hereditary optic neuropathy patients, observed that only 11.5% had onset of the disease in childhood; among these, patients with acute bilateral Leber hereditary optic neuropathy (as in our case) had a mean age of onset of 7.4 ± 3.2 years. The mean age at disease onset is approximately 10 years in the Chinese population.2,4,12
In our review of the literature on pediatric female children, the mean age at symptom onset was 11 years, probably because of the predominance of Asiatic literature reporting a more advanced onset age in pediatric females. Only 4% of patients had symptom onset at preschool age. An onset at age as young as 3 years in female children has been described in only 3 cases in the literature, including our patient. We defined it as “very early onset.”
Very early onset Leber hereditary optic neuropathy is rare because significant complex I dysfunction is required to cause mitochondrial failure and subsequent optic nerve damage, findings that usually appear later in life. 103
Disease onset at a very young age may result from genetic variants leading to an increased risk of visual loss or to an increased susceptibility to environmental triggers. 105 However, the variant described in our child is among the 3 most common disease-causing genomic variants of Leber hereditary optic neuropathy. We therefore hypothesize that in our patient, Leber hereditary optic neuropathy was caused by modulation of genetic and/or epigenetic factors, yet unidentified, negatively modulating the phenotype or by the occurrence of heteroplasmy and mitotic segregation.102,106
Our review of the literature highlights that the clinical features at onset are rarely described in female children, available only in 40.5% of the patients, since the focus in the literature is mainly on genetic characterization.
The onset modality of Leber hereditary optic neuropathy was not specified for approximately half of the female children reported in the present review. Acute onset was the most frequent mode of presentation, characterized by sequential bilateral visual loss.1,4–6,12–60 Visual loss was mild or moderate in the majority of cases.4,13,104 Fundus oculi examination was available in only 16% of the cases, and the most frequent presentation was acute with edema of the optic disc.
In very young children, visual loss may be undetected when it is monocular or not severe; moreover, the diagnostic tests available in adults for the characterization of visual loss are difficult to perform in young patients. In the first case with very early onset, clinical information was scarce consisting in the report of visual impairment at onset only. 21 The second case was a female presenting with exotropia and bilateral visual impairment (left eye more than right eye): her fundus examination at onset revealed bilaterally pale optic disc, visual evoked potentials were similar to ours, and she never recovered. 28
The presentation of very early onset Leber hereditary optic neuropathy is challenging for pediatricians, particularly if it includes pseudo-optic disc edema.
Indeed, the differential diagnosis of acute and subacute vision loss could be due to a variety of medical conditions, including optic neuritis and other forms of optic neuropathy. 7
In the presence of optic disc edema, the first differential diagnosis is between raised intracranial pressure and the condition of pseudo-optic disc edema (ie, the elevation of the optic disc in the absence of intracranial hypertension). The differential diagnosis requires brain and spinal magnetic resonance imaging to search for the presence of mass lesions, cerebral sino-venous thrombosis, and the nonspecific indicators of hydropathic intracranial hypertension (posterior globe flattening, distention of the cerebrospinal fluid space, empty or partially empty saddle, and transverse venous sinus stenosis). 107 In patients with normal MRI, lumbar puncture may be used to evaluate the opening pressure. 7
The second important differential diagnosis is pseudo-optic disc edema, a condition that includes optic nerve drusen, optic neuritis, and other optic neuropathies. Other diagnostic tools, such as visual field examination, optical coherence tomography, optic bulb ultrasonography, and visual evoked potentials are required to diagnose the conditions underlying pseudo-optic disc edema.8,108,109 Our literature search retrieved very few data based on these diagnostic tools, probably because they are difficult to perform and interpret in young children.
Among the differential diagnoses of Leber hereditary optic neuropathy, the most challenging is anterior optic neuritis. Clinically, pain during eye movement, dyschromatopsia, and afferent pupillary defects suggest optic neuritis. 109 However, the afferent pupillary defects are seldom seen in pediatrics because optic neuritis is usually bilateral and pain or dyschromatopsia are difficult to evaluate at a young age. 7 In these cases, visual evoked potentials can be useful even in very young children. In acute Leber hereditary optic neuropathy, they show amplitude decrease and delayed latency of the P100 peak. Shortening of P50 is an additional feature in those with a history of Leber hereditary optic neuropathy longer than 3 months. 104 In contrast, in optic neuritis, visual evoked potentials show a predominant delay in the latency of the response, whereas amplitude is more variably affected. The most important differential feature is visual evoked potentials recovery following steroid therapy in optic neuritis but not in Leber hereditary optic neuropathy.104,109
The diagnosis of Leber hereditary optic neuropathy needs to be confirmed by detection of disease-causing genetic variants in mitochondrial DNA; methods such as direct DNA sequencing, polymerase chain reaction, restriction fragment length polymorphism, and massive parallel sequencing can be used to identify mitochondrial DNA genetic variants. 110
The diagnosis was a challenge in our case because the family history was uneventful since the mother and sister were healthy carriers of the same mitochondrial disease-causing genetic variant. 103
The natural history of Leber hereditary optic neuropathy is characterized by progressive loss of visual function, although the advent of Idebenone treatment was associated with recovery or stabilization in 3 of 6 patients.28,40 In the present study, the outcome of Leber hereditary optic neuropathy is reported in 27% of female children. Approximately half of them had visual recovery or stabilization of vision, and only a minority received Idebenone (10%) or gene therapy (16%), with variable success. Early therapy has been reported to arrest the progression of visual disease9–11; Idebenone was approved in 2015 by the European Union. Its use before 12 years of age is off-label.16,111 In our patient, Idebenone treatment was started before the evolution from optic disc edema to optic disc atrophy, and visual function did not deteriorate during the following 5 years.
The main limitations of the present study are the heterogeneity of the collected patients and the incomplete information on some clinical data (recovery, type of onset, and neuro-ophthalmologic information). The limit of the exclusion of pediatric-onset Leber hereditary optic neuropathy studies in which sex was not specified75–80 or which considered only males69–74 would not have influenced the estimate of the pediatric male-to-female ratio because of the low number of only male patient studies. We did not perform a double-blind revision of collected data during literature review. However, this is the first study reporting specific clinical information on the population of pediatric females affected by Leber hereditary optic neuropathy. Increasing clinical awareness of the rarest phenotypes of Leber hereditary optic neuropathy, such as that of young females, has important implications for early diagnosis and effective management.
Conclusion
Leber hereditary optic neuropathy in female children, particularly with early pediatric onset, is extremely rare. Only 3 cases, including ours, are described with an onset as early as 3 years. Child neurologists facing very young female children with acute-subacute visual loss, bilateral pseudo-optic disc edema, and visual evoked potential changes not responding to steroid therapy should consider Leber hereditary optic neuropathy early in the course of disease after excluding other more common causes. Early and timely diagnosis is important to preserve vision.
Supplemental Material
sj-doc-1-jcn-10.1177_08830738221149962 - Supplemental material for Leber Mitochondrial Optic Neuropathy in Pediatric Females With Focus on Very Early Onset Cases
Supplemental material, sj-doc-1-jcn-10.1177_08830738221149962 for Leber Mitochondrial Optic Neuropathy in Pediatric Females With Focus on Very Early Onset Cases by Sara Tagliani, MD, Cristina Malaventura, MD, Chiara Ceccato, MD, Francesco Parmeggiani, MD, PhD, and Agnese Suppiej, MD, PhD in Journal of Child Neurology
Footnotes
Acknowledgments
We thank the ophthalmologists Dr Cermakova and Dr Maritan and the orthoptist Roberta Guerriero for sharing the clinical data, the psychologists Tiziana Battistin for the cognitive evaluation, and Caterina Paderni for support during the ophthalmologic evaluations and the multidisciplinary follow-up.
Author Contributions
ST contributed to conception and design and wrote the first draft of manuscript. CM contributed to conception and design of the study, interpretation of the data and revised the manuscript for important intellectual content. CC contributed to data acquisition and interpretation, and drafted ophthalmologic data. FP contributed to interpretation of ophthalmologic results and critically reviewed the manuscript. AS conceived and designed the study, supervised the interpretation of the data, wrote the final version of the manuscript. All authors approved the final version.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Ethical Approval
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.