
Abstract

Dear Editor,
We read with great interest the article titled “Rare Cases of Pseudomonas aeruginosa Meningitis in Children: 10-Year Experience in a Single Center” recently published in your journal. 1 The authors’ comprehensive analysis of 10 pediatric patients diagnosed with Pseudomonas aeruginosa meningitis provides valuable insights into the clinical characteristics and outcomes associated with this rare but devastating condition. We were particularly intrigued by the reported predispositions, such as prior neurosurgical procedures (in 3 patients) and underlying leukemia (in 4 patients). However, the article prompted us to consider whether immunologic evaluation should be pursued more broadly in children presenting with P aeruginosa meningitis, especially in those without obvious predisposing conditions.
Herein, we would like to share our experience involving a previously healthy 7-month-old female infant who succumbed rapidly to P aeruginosa meningitis and was ultimately found to have an underlying inborn error of immunity—MYD88 deficiency (NM_002468.5:c.98T>C, p.Leu33Pro). This case not only adds to the growing recognition of pathogen diversity in primary immunodeficiencies but also highlights the potentially fatal course of P aeruginosa infections in infants with impaired innate immunity.
The patient presented to our emergency department with a 3-day history of fever, cough, and lethargy. On admission, she was unconscious (Glasgow Coma Scale score of 7) and required immediate intubation and transfer to the pediatric intensive care unit. Her immunizations were up-to-date, and there was no history of prior hospitalizations or infections. However, her family history was notable for consanguinity and the death of a sibling from meningitis at 18 months of age.
Neurologic examination revealed signs of raised intracranial pressure, including diminished pupillary light reflexes, mid-dilated pupils, and a tense anterior fontanel. Cranial MRI revealed restricted diffusion in the anterior part of the left lateral ventricle extending to the lentiform nucleus, indicating severe meningeal inflammation (Figure 1).
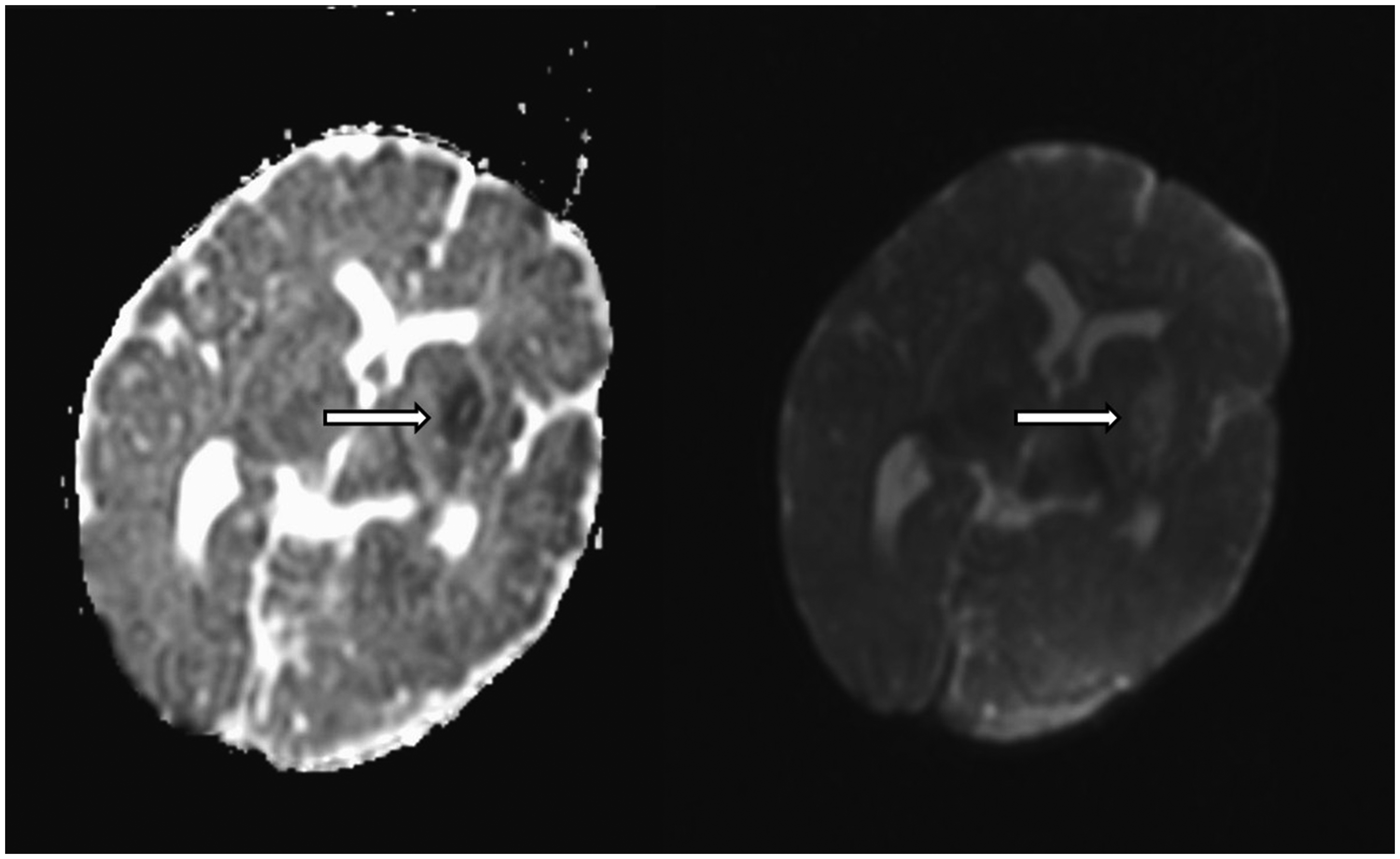
Brain Diffusion-Weighted MRI of the Patient Showing Restricted Diffusion in the Anterior Horn of the Left Lateral Ventricle and Lentiform Nucleus, Consistent With Severe Meningeal Inflammation.
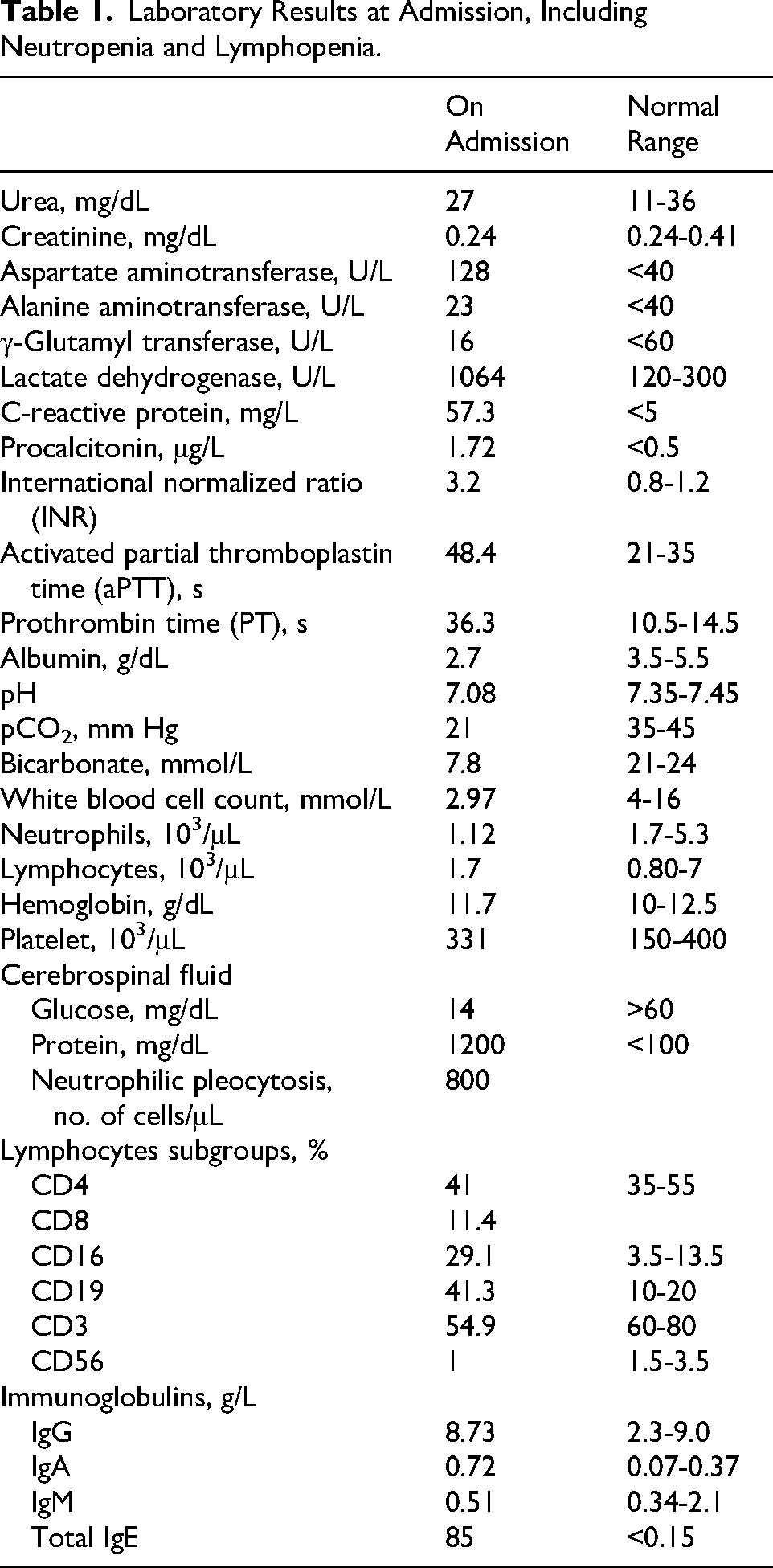
Cerebrospinal fluid analysis showed profoundly decreased glucose (14 mg/dL), elevated protein (1200 mg/dL), and marked neutrophilic pleocytosis (800 cells/μL). Simultaneously, the patient exhibited both neutropenia (absolute neutrophil count: 1.12 × 10³/μL) and lymphopenia (absolute lymphocyte count: 1.7 × 10³/μL), as shown in Table 1. These findings suggested underlying immunologic impairment beyond acute infection–related cytopenia.
Laboratory Results at Admission, Including Neutropenia and Lymphopenia.
Empirical antibiotic treatment with ceftriaxone and vancomycin was initiated, later escalating to meropenem and amikacin because of clinical deterioration. Cerebral edema was managed with hypertonic saline, and fresh frozen plasma was administered for coagulopathy. Despite aggressive resuscitation efforts, including high-dose vasoactive support and intravenous immunoglobulin administration, the patient developed refractory septic shock and suffered a cardiac arrest 47 hours after admission.
Postmortem blood and cerebrospinal fluid cultures yielded Pseudomonas aeruginosa, sensitive to meropenem and amikacin. Genetic testing performed 2 months later identified a homozygous MYD88 variant NM_002468.5:c.98T>C (p.Leu33Pro). This variant was absent from gnomAD (allele frequency = 0), predicted deleterious by SIFT, probably damaging by PolyPhen-2, and had a high CADD score. According to VarSome, the variant was classified as pathogenic (ACMG criteria PM1, PM2, PP3). Additionally, parental screening revealed heterozygosity for a CARD11 variant. Although CARD11 deficiency was considered because of parental consanguinity and the death of a sibling, the clinical phenotype of our patient (early-onset, invasive bacterial infection) was not consistent with CARD11 deficiency.2,3 Thus, extended testing including severe combined immunodeficiency and innate immunity genes was requested, which led to the identification of the MYD88 variant.
MYD88 deficiency is a rare autosomal recessive disorder that disrupts Toll-like receptor and IL-1 receptor signaling pathways, leading to impaired activation of NF-κB and defective cytokine responses. Although encapsulated organisms such as Streptococcus pneumoniae and Staphylococcus aureus are the most commonly implicated pathogens, our case underscores the broader infectious spectrum that includes P aeruginosa. Although gram-negative infections in MYD88-deficient patients are infrequent, they have been reported in recent literature and are often associated with severe clinical outcomes.4–6
In the study by Von Bernuth et al, 2 8 patients with MYD88 mutations experienced life-threatening bacterial infections, predominantly involving Pneumococcus and Staphylococcus. Rudilla et al 3 further emphasized pathogen variability in primary immunodeficiencies, cautioning against underestimating the role of gram-negative organisms. Our case is consistent with these findings and expands the clinical phenotype of MYD88 deficiency to include fulminant P aeruginosa meningitis in infancy. Furthermore, early recognition of MYD88 or IRAK-4 deficiency has been shown to allow preventive strategies such as prophylactic antibiotics and intravenous immunoglobulin, which may reduce invasive infections.6,7
We believe this case highlights the importance of considering primary immunodeficiencies in infants with severe or atypical bacterial infections, particularly when accompanied by cytopenias or a concerning family history. Although neutropenia and lymphopenia can be reactive in sepsis, their presence at admission in our patient—along with abnormal lymphocyte subsets (eg, low CD3 and CD56 expression)—provided early clues suggestive of an underlying immunologic disorder. In our patient, lymphopenia was observed; however, MYD88 deficiency is primarily known to affect TLR/IL-1R signaling, leading to susceptibility to pyogenic bacterial infections. Interestingly, in another reported case of MYD88 deficiency, lymphopenia and abnormalities in lymphocyte subsets were not observed. 7
In interpreting this case, certain limitations should be acknowledged. Segregation analysis of the MYD88 variant could not be performed, as parental and sibling testing at the time was limited to severe combined immunodeficiency–associated genes. Following the patient's death, the family declined additional genetic investigations, precluding further confirmation. Furthermore, functional validation of the variant, such as cytokine release after lipopolysaccharide stimulation, was not feasible because the patient died within 3 days of admission.
In conclusion, we commend the authors for drawing attention to this rare but important cause of pediatric meningitis. In light of our findings, we propose that comprehensive immunologic and genetic evaluation be considered in all pediatric cases of P aeruginosa meningitis, even in the absence of overt risk factors. Such an approach may facilitate early diagnosis of primary immunodeficiencies, guide targeted therapies, and potentially improve outcomes in this vulnerable population.
Footnotes
Ethical Approval
Since this is a case report, ethical committee approval was not required according to our institution's guidelines, and informed consent was obtained from the patient.
Author Contributions
All authors reviewed and approved the final manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.