
Abstract
Glucose transporter type 1 deficiency syndrome (GLUT1DS) is a genetic condition associated with complex neurologic symptoms, including epilepsy. Ketogenic diet therapy (KDT) is considered the standard treatment for GLUT1DS. This retrospective study identified trends in treatment with KDT for patients with GLUT1DS to optimize the current standards of care.
Methods
A retrospective chart review was performed to identify patients at a pediatric institution with GLUT1DS receiving the ketogenic diet.
Results
Twelve patients were identified; 10 met inclusion criteria. A classic ketogenic diet (cKD) with a 3:1 ratio provides effective support for patients in this sample.
Conclusion
Results of the study suggest that a 3:1 ratio of KDT, which may increase tolerance and adherence and reduce adverse effects, may be acceptable in patients with GLUT1DS.
Introduction
Glucose transporter type 1 deficiency syndrome (GLUT1DS) is characterized by impaired transport of glucose to the brain because of the impact on glucose transport type 1 (GLUT1) proteins.1,2 GLUT1DS is an autosomal dominant genetic disorder that is commonly associated with SLC2A1 pathogenic variants. The resulting disturbance in the transport of glucose across the blood-brain barrier causes an energy deficiency, and ultimately, a broad clinical spectrum of complex neurologic symptoms including developmental delay, progressive microcephaly, abnormal movements, and seizures. 2 Seizures are a common symptom of GLUT1DS and often begin in infancy and progress to become intractable. 3 Seizures may be the first clinical sign of GLUT1DS as they often begin before 6 months of age. Individuals with GLUT1DS typically experience seizures with generalized onset such as typical and atypical absence, myoclonic, atonic, and generalized tonic clonic, with focal seizures being less common. 4 Absence seizures that begin younger than age 4 are a known characteristic of GLUT1DS. Regardless of seizure semiology, ketogenic diet therapy (KDT) is known to improve seizure control.
GLUT1DS is associated with hypoglycorrhachia and normoglycemia. When suspected, fasting cerebrospinal fluid (CSF) glucose should be obtained. Typically, the brain uses glucose for metabolic energy; however, because of the disturbance in glucose transport to the brain in GLUT1DS, an alternative form of energy is necessary to promote brain function. 2 It is important to recognize and diagnose early as GLUT1DS can be treated with a KDT.
KDT is the standard treatment for GLUT1DS and is recommended to start as early in life as possible.1,2 KDT consists of a high-fat, low-carbohydrate, and adequate-protein diet that is prescribed in a ratio of grams of fat to grams of carbohydrate and protein combined. A standard diet uses carbohydrates for energy; however, in KDT, the body uses fat for energy. This process mimics starvation. When glucose is not available for metabolism, the body uses fat for energy. The use of fat for energy results in the production of ketone bodies. These ketone bodies act as an alternative source of energy for the brain in patients with GLUT1DS. This is necessary not only for brain nourishment but also for neurodevelopment and seizure control. Ketones serve to protect against seizures through multifactorial and often debatable mechanisms. 5
Ketogenic diet therapy remains the standard of care in those with GLUT1DS, with therapy being initiated as early as possible with the highest degree of ketosis tolerated. 2 Generally, ketones can be measured in blood and urine. According to the recommendations by the international GLUT1DS study group, β-hydroxybutyrate (BHB) levels should be monitored either in capillary or venous blood. 2 For patients on KDT, the ratio of fat to carbohydrate and protein combined may be altered based on levels of ketosis, efficacy, and adverse effects experienced while on diet therapy. Choice of KDT is often influenced by the patient’s age and tolerability, with intolerance contributing to eventual discontinuation in some patients because adverse effects of this diet may alter the patient's quality of life. 2 The risk of adverse effects increases on higher ratios of KDT.2,6 Adverse effects such as dehydration, electrolyte abnormality, hypoglycemia, acidosis, constipation, and other gastrointestinal disturbances may occur at the time of diet initiation or months after starting diet therapy. 7 Long-term effects may include elevated serum lipids, kidney stones, and impaired bone health. 7 The adverse effects are recognized from parental reports as well as from routine laboratory work. Patients who are initiated on KDT should have regular follow-up at 1, 3, 6, 9, and 12 months post initiation with laboratory work obtained with each of those visits.8,9 Adverse effects are typically reversible and easily corrected if patients are followed closely by experienced ketogenic diet teams.
Currently, there are limited studies that define specific therapeutic BHB ranges or show the efficacy of higher ratio versus lower ratio ketogenic diet therapies for the treatment of GLUT1DS.10–13 It is recommended that under the age of 2 years, classic KDT is the treatment of choice while adolescents and adults may benefit from lower ratios or Modified Atkins Diet (MAD) for quality of life and compliance. 2 There is a lack of formal recommendations in ratio of KDT or degree of ketosis for children between the ages of 2 and adolescence. In this age group, tolerability of KDT for continuation of therapy must be considered. There is also a lack of formal recommendations in ratio and degree of ketosis for those with GLUT1DS. As therapy is often initiated as high as tolerated, these patients may be maintained on higher ratios of the KDT than necessary. A survey of primarily experienced registered dietitian nutritionists (RDNs) from 34 centers among 10 countries who carry out training of KDTs in children with GLUT1DS, reported a preference for implementing a KDT using carbohydrate counting or a classic ketogenic diet (cKD) ratio lower than a 3:1. 14
This retrospective study aims to identify trends in treatment with KDT in patients with GLUT1DS to contribute to the literature and identify potential changes in practice to current standards.
Patients and Methods
A retrospective chart review was performed to identify patients at a large urban pediatric institution in the Midwest United States who had been diagnosed with GLUT1DS between June 2008 and June 2023. Inclusion criteria consisted of a diagnosis of GLUT1DS and treatment with the classic ketogenic diet. All patients met diagnostic criteria for GLUT1DS with developmental delay, seizures, hypoglycorrhachia, and normoglycemia or a pathogenic SLC2A1 gene mutation. Treatment protocol for cKD initiation involved a 4-day hospital admission, with advancement from a 1:1 to 2:1 to 3:1 ketogenic ratio. Blood glucose, urine ketones, and clinical symptoms were monitored during the initiation period. Diet education was provided daily at the bedside. Initially, 12 patients were identified. One patient was excluded because of parental refusal to start KDT. One additional patient was excluded whose diet therapy was initiated at an outside institution, and thus initiation data was unknown. The resulting final sample for this review was 10 patients.
Data collected included the age of GLUT1DS diagnosis and age at cKD initiation. The ratio of the diet at initiation and the ratios at 1 month, 6 months, and 1 year were recorded. The venous BHB level associated with those times of 1 month, 6 months, and 1 year were also documented, per the standard of care protocol at our institution. Seizure frequency data and data related to seizure medication requirements were collected at initiation and at 1 year. Adverse effects of the diet necessitating supplementation were also documented.
Results
There was a total of 12 patients identified as having GLUT1DS; 2 patients were excluded for not meeting inclusion criteria. The remaining patients consisted of 4 males (40%) and 6 females (60%). The mean age at diagnosis was 3.75 years (range 3 weeks to 16 years). All 10 patients had genetic testing with a mutation identified in the SLC2A1 gene. Cerebrospinal fluid (CFS) testing was performed in 4 of 10 (40%) of the patients, all of which showed hypoglycorrhachia. Development at diagnosis was noted to be normal in 2 of 10 (20%) of the patients with some degree of developmental delay noted in 8 of 10 (80%). Abnormal movement outside of seizures was noted in 6 of 10 patients (60%), with abnormal eye movements and ataxia being the most common abnormal movements noted. The mean age at ketogenic diet initiation was 4.03 years. Baseline seizure frequency was available. For the 10 patients included in the review, 5 had rare seizures, 1 had a few seizures per month, 3 had daily seizures, and 1 was seizure free. For the 10 patients, 10 of 10 (100%) required antiseizure medications (ASMs) at the time of diet initiation. Five patients were on levetiracetam monotherapy, 1 was on levetiracetam and valproic acid, 1 was on levetiracetam and ethosuximide, 1 patient was on valproic acid monotherapy, and 1 was on clobazam monotherapy (Table 1).
Patient Demographics.
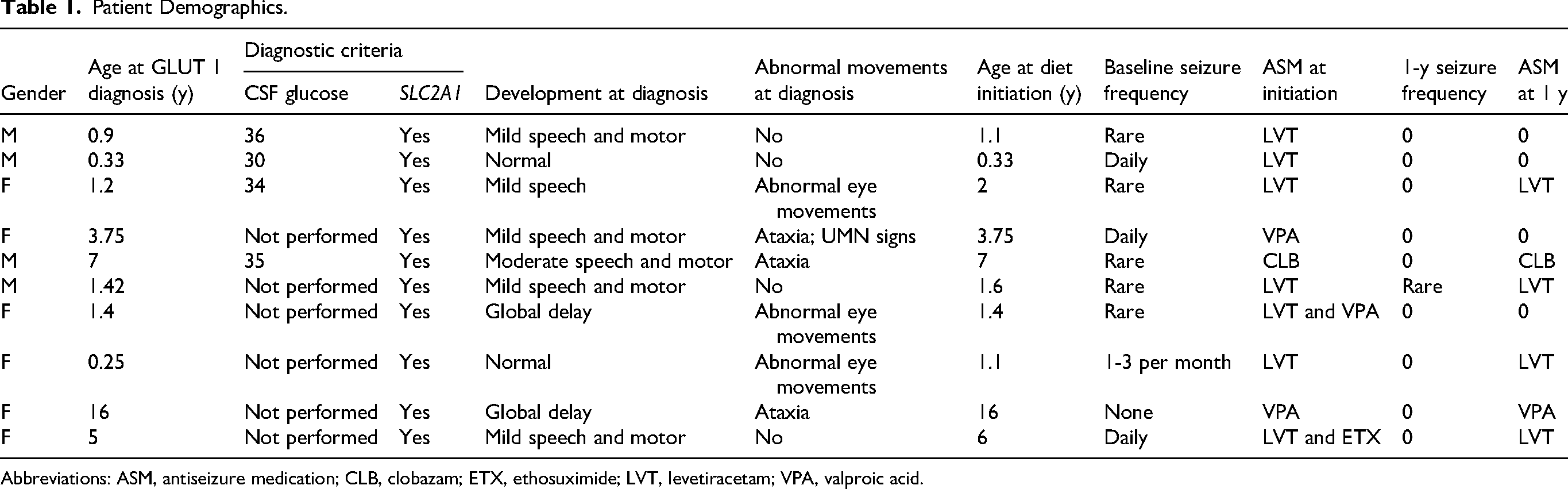
Abbreviations: ASM, antiseizure medication; CLB, clobazam; ETX, ethosuximide; LVT, levetiracetam; VPA, valproic acid.
One-year follow-up data for diet ratio was available for 6 of the 10 patients, 4 of 6 (67%) were on a 3:1 ratio and 2 of 6 (33%) were on a 2:1 ratio (Figure 1). Mean BHB level was 3.5 at 1 month, 3.6 at 6 months, and 3.5 at 1 year. At 1 year, 4 of 10 (40%) were off all ASMs and 6 of 10 (60%) were still on 1 ASM (1 on clobazam, 4 on levetiracetam, 1 on valproic acid). Of the 6 patients requiring an ASM at 1 year, none required polypharmacy. Parentally reported subjective data on cognition and behavior was elicited at each follow-up ketogenic diet clinic visit. At the 1-year visit, 7 of 10 parents (70%) reported improvements in cognition and behavior, 1 reported improved cognition but no change in behavior, and 2 reported no noted change in cognition or behavior.
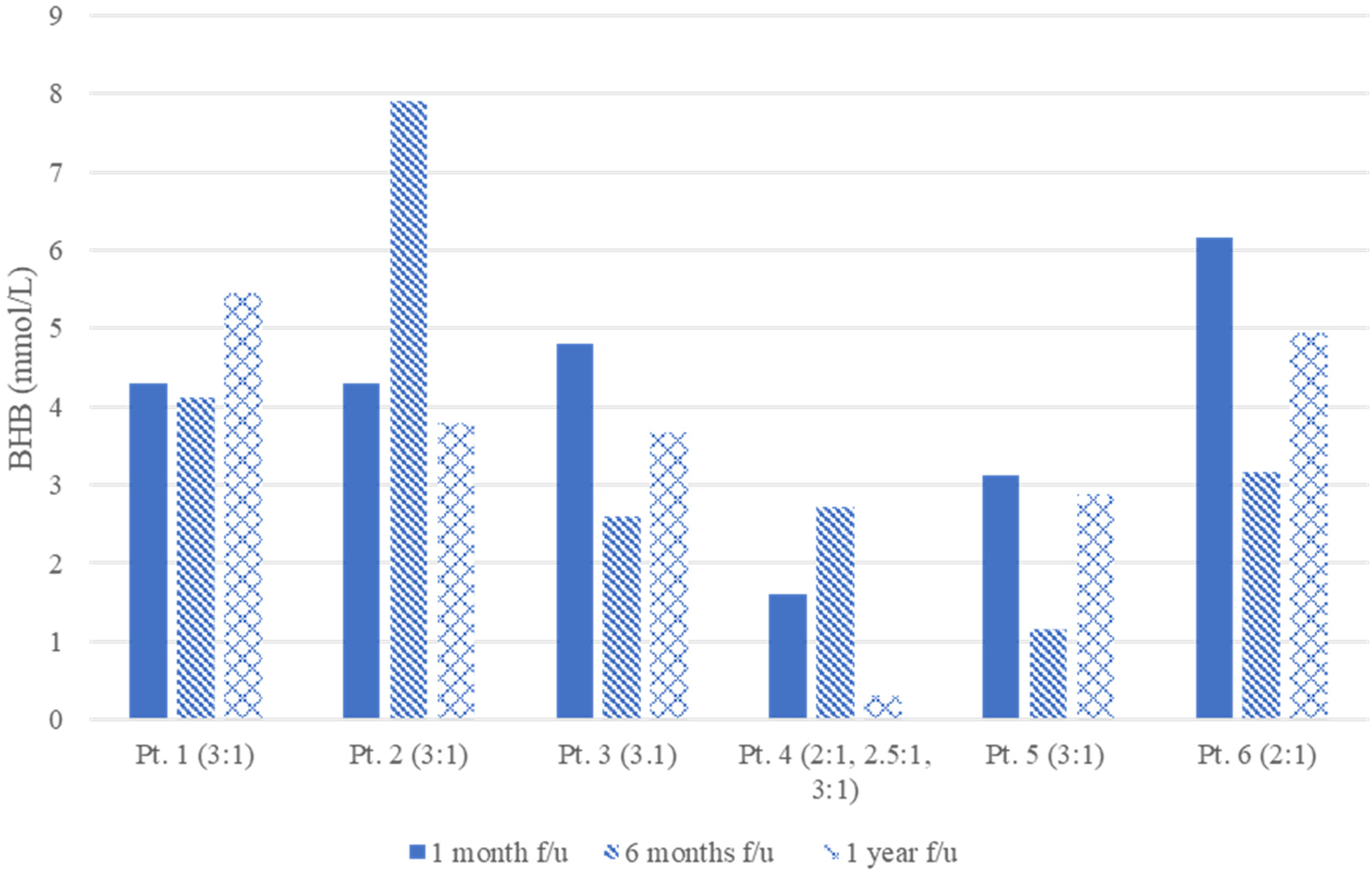
β-Hydroxybutyrate (BHB) levels with associated ratio at follow-up.
Adverse effects of KDT requiring supplementation were observed in this cohort. Carnitine deficiency was observed in 80% of the patients, thus requiring levocarnitine supplementation. Metabolic acidosis, defined by a serum CO2 level of less than 18 mmol/L at our ketogenic diet center, required treatment of sodium bicarbonate and/or potassium bicarbonate in 4 of 10 patients (40%). Constipation requiring treatment with polyethylene glycol laxative powder was observed in 2 of 10 patients (20%).
Discussion
Multiple studies have confirmed the efficacy of KDT for use in people with epilepsy and specifically have demonstrated that it is the gold standard for patients with GLUT1DS.2,3,14–16 Prior studies lacked consistent definitions for target fat-to-nonfat ratios or BHB levels specifically for people with GLUT1DS that require long-term treatment with KDT. Because of a lack of consistent recommendations in these areas, providers may opt to maximize the ratio of the diet to ensure the highest level of ketosis is achieved.
This study identified 10 pediatric patients with a mean age of 4.5 years at ketogenic diet initiation. Patients were able to maintain a mean BHB of 3.5 to 3.6 mmol/L across the first year of diet therapy on a mean fat-to-nonfat ratio of 3:1. Sixty percent of these patients also used medium-chain triglyceride oil, or MCT oil, as part of their cKD in varied daily amounts. MCTs are quickly absorbed into the bloodstream and sent directly to the liver via the portal vein, where they are rapidly oxidized to produce ketones. MCT oil is commonly used as part of a ketogenic diet in this patient population and in our practice. It is possible that because a majority of this cohort took MCT oil with their cKD that this allowed them to remain in stronger levels of ketosis even at lower ketogenic ratios.
At diet initiation, all patients required ASM, with all patients experiencing some level of seizure activity. The diet's clinical effectiveness over time was evaluated using seizure frequency and ASM requirement outcomes. At 1 year of diet therapy, 4 of 10 patients no longer required ASM, and those who did required treatment with a single ASM in addition to KDT, suggesting that polypharmacy was not necessary to promote seizure control. The KDT may influence plasma levels of ASMs, or the combination of KDT and ASMs may exacerbate potential side effects. 17 Regular drug monitoring and laboratory studies are necessary for ongoing comprehensive care. Although a small cohort, the data on ASMs are valuable given there is limited knowledge about which drugs may be effective in combination with KDT for patients with GLUT1DS.
Adverse effects related to the classic ketogenic diet were relatively mild, which was likely due to the lower diet ratios. None of the patients were removed from the diet because of intolerable side effects. Carnitine deficiency was mitigated with levocarnitine supplementation. The international GLUT1DS group noted that routine carnitine supplementation is a subject of debate. Although most centers regularly monitor serum carnitine levels to address deficiencies, some choose to provide levocarnitine empirically to patients on ketogenic diet therapy. 2 The standard of care at our institution is to supplement levocarnitine for those that show a deficiency after the start of KDT. Acidosis was monitored closely and corrected effectively with potassium bicarbonate, potassium citrate, or sodium bicarbonate. Only 2 of the patients reported constipation and used polyethylene glycol. There were no reported cases of kidney stones in our cohort; whereas the literature suggests a 3% to 7% occurrence in children maintained on KDT. 8 Relative to clinical impact, ketosis level, and tolerability, results of this retrospective study suggests that fat-to-nonfat ratios higher than 3:1 on ketogenic diet therapy may not be necessary for adequate ketosis and effective management of GLUT1DS.
Limitations
The clinical neurologic impact of adequate ketosis in this review is limited to seizure control alone, and further studies are warranted to determine the clinical impact of ketogenic diet therapy on other neurologic factors associated with GLUT1DS. Retrospective data collection limited data availability for all patients within the cohort. Data collection was limited to 1 year after initiation; therefore, data did not reflect long-term adherence, clinical impact, or side effects. This review considered carnitine deficiency, metabolic acidosis, and constipation; however, future investigation is warranted to consider long-term clinical impact and side effects including bone health and kidney stones. Although this study supported a lower ratio for KDT, it did not track long-term adherence to the diet. Other studies have suggested that higher ratios of KDT may negatively impact adverse effects and compliance. Longitudinal tracking of patient adherence is necessary to determine if higher ratios impact long-term compliance.
The cohort in this review represented a small sample size from a single site. Additional studies with a larger cohort size are warranted to determine broader impact and consistency of data. However, this review contributes evidence in support of lower ratios of ketogenic diet therapy for patients with GLUT1DS.
Conclusion
This study found that a 3:1 ratio of grams of fat to grams of nonfat in the cKD was effective in GLUT1DS patients maintained on ketogenic diet therapy. The mean BHB across the first year of therapy was maintained at 3.5 to 3.6 mmol/L and the need for ASMs was reduced. Those who continued to need an ASM were predominantly treated with levetiracetam. Most patients required levocarnitine supplementation; however, acidosis and constipation were less of a concern. None of the patients stopped the diet because of tolerance or side effects. Results of the study suggest that a 3:1 ratio of KDT may be acceptable in patients with GLUT1DS and may aid in mitigating side effects of long-term use of the diet. Larger studies are recommended to further evaluate and establish standardization of ketogenic diet ratios in patients with GLUT1DS for seizure control and goal range of BHB levels.
Footnotes
Ethical Considerations
This study received initial ethical approval from the Lurie Children's institutional review board (IRB; approval #2018-1978) on May 14, 2018, with its most recent renewal on September 30, 2025. This is an IRB-approved retrospective study; therefore, all patient information was deidentified, and patient consent was not required. Patient data will not be shared with third parties.
Consent to Participate
The IRB also waives the requirement of obtaining informed consent for this study for another approval period in accordance with 45 CFR 46.116(d): (1) the research involves no more than minimal risk to subjects; (2) the waiver or alteration will not adversely affect the rights and welfare of the subjects; (3) the research could not practicably be carried out without the waiver or alteration; (4) whenever appropriate, the subjects will be provided with additional pertinent information after participation.
Consent for Publication
Not applicable.
Author Contributions
BF and RB designed the study and coordinated the ethical approval process. BF, RB, SM, and WL participated in data collection. BF led the interpretation of findings. All authors drafted, reviewed and revised the manuscript and approved the decision to submit for publication.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.