
Abstract
Autism spectrum disorder (autism) is diagnosed by persistent deficits in communication and social interaction, along with restricted, repetitive behaviors or interests. About a third of children with autism appear to develop normally but subsequently regress and eventually present with autism. This condition is termed regressive autism and the associated regression termed autistic regression. Children undergoing autistic regression after 2 years are described as having childhood disintegrative disorder. We aimed to conduct a scoping review to identify and summarize the genetic etiologies and correlates of regressive autism and childhood disintegrative disorder. Using key words, we searched 4 databases for papers published from January 2010 to February 2024. Thirty-two papers were retained. Nearly 90 genetic variants were associated with these conditions, and some treatments improved proband functioning. Epigenetic involvement, immune dysfunction and toxicant exposures were related to autistic regression while recurring comorbidities were inflammatory bowel disease, fever and mitochondrial disease. Regressive autism is variably defined, impeding research. The development of a precise definition is needed. Furthermore, regressive autism and its subtype, childhood disintegrative disorder, have many causes, which means that developing biomarkers and endotypes to explore etiologies would likely pay dividends. Because of the increasing prevalence of autism, regressive autism is no longer a rare condition, emphasizing the grave need to promote research in this area. Expected benefits might be improved outcomes for those affected and genetic counselling for at-risk family members. Longer-term benefits might be reduced prevalence, less emotional and financial burdens for families, and lower fiscal burdens for governments.
Autism spectrum disorder (hereafter autism) is a diverse neurodevelopmental disorder with symptoms mostly observed before 2 years. 1 Children exhibiting persistent deficits in social communication and interaction, along with restricted and repetitive behaviors, interests or activities, 1 qualify for an autism diagnosis. Etiology is heterogeneous with a strong genetic contribution. 2 Autism prevalence in 8-year-olds in the United States has risen from 0.4% in 2000 to 3.22% in 2022 3 with no established cause for the more than 700% increase. This review focuses on autism subtypes involving regression.
Autistic regression is the loss of acquired skills such as speech, social skills, and toileting. 4 About 1 in 3 children with autism have experienced regression, largely of unknown origin. 5 When used as an umbrella term, regressive autism (RA) includes all children who have had autistic regression, here termed broader regressive autism. However, RA can also be used to describe children who have suffered regression before a particular age. Here, regression before 2 years is termed RA. Compared to children with early-onset autism (EOA), those who have experienced regression, tend to have more severe symptoms and a worse prognosis. 6 Childhood disintegrative disorder (CDD), another subtype of broader RA, is diagnosed in children who have had 2 or more years of apparently normal development with subsequent autistic regression. 7
Because of its rarity, CDD prevalence is difficult to estimate, but a researcher in the United States 8 estimated prevalence as 2.0 per 100 000. The Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, 9 included CDD as a distinct diagnosis but it was subsumed by autism in the subsequent and fifth edition. 1 Hence, some children with CDD are formally diagnosed only with autism. Similarly, RA is not a distinct diagnosis in the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5), so again, the diagnosis may only be autism with no regression mentioned. These factors understate the prevalence of RA and CDD and make them even more difficult to research. In this review, broader RA groups the subtypes RA and CDD.
Disorders such as autism are diagnosed by behavioral symptoms, and the labels RA or CDD are sometimes added by a clinician after considering a patient's developmental history. Contrastingly, a plethora of disorders are diagnosed unequivocally via blood and other tests. Behavioral diagnoses are wide-ranging and sometimes imprecise, resulting in no clear, consistent etiologies for RA or CDD. Other than mitochondrial defects, no definitive etiologies have been established for broader RA. 10
A small subset of children with broader RA has early developmental delays and then regress. This group aside, the development of children with broader RA appears normal before regression, hinting that causal factors are different from EOA. Thus, researching etiologies and associations of broader RA, separately from autism, is critical.
Recently, it was estimated that rare genetic variants are causal, or at least contributory, in 10% to 30% of people with autism, a large increase over the last 15 years when a genetic cause could only be discerned in 2% to 3%. 11 This indicates a vital need to review, compile and disseminate genetic and other information related to RA and CDD to researchers, clinicians and affected families.
Because of the limited research and understanding of RA and CDD,4,12 it is particularly important to gather data for these subtypes of autism. Improving understanding of their genetic and other associations could lead to improved prevention, diagnoses, and treatments whilst also increasing our knowledge of EOA and enabling better genetic counselling for affected families. Care of children with broader RA is expensive as most need high-level, lifelong support. Hence, improving the functioning of these children and/or reducing the prevalence of broader RA would lessen the emotional and financial burden of families and the fiscal burden of governments.
To date, we have found no comprehensive review aiming to identify genetic and other risk factors of broader RA. Via this scoping review, we will be enabled to map the available research in this area and identify any gaps. 13 Hence, this was our chosen method to achieve our aim of identifying genetic and other risk factors of RA and CDD.
Methods
Registration of the Review
This review was submitted for registration to Prospero on 8 January, 2024 (CRD42023468974).
Search Strategy
We systematically searched PubMed, MEDLINE, Embase and ProQuest Dissertations and Theses Global (hereafter ProQuest) databases for studies published from January 2010 to 29 February 2024, inclusive. For PubMed, MEDLINE and Embase, our search terms, existing anywhere in the article, were (‘disintegrative disorder’ or ‘regressive autism’ or ‘autistic regression’) and (‘epigenetics’ or ‘gene’ or ‘genetics’ or ‘genomics’ or ‘histone modification’ or ‘DNA methylation’). Because of the increased length of dissertations and theses in ProQuest, we only searched abstracts of these whilst using the same strategy.
Eligibility Criteria
An abstract/article was included if it (1) reported original research or was a review, (2) was available as English full-text, (3) was from a peer-reviewed journal or a PhD thesis/dissertation, and (4) related to our objective of identifying and summarizing genetic and other risk factors of RA and CDD. Because of the known scarcity of research into RA and CDD, all study types were included. Abstracts (or articles where no abstract) identified by searches were screened independently by 2 co-authors (J.F., F.R., A.G. or J.V.). Where reviewers’ assessments differed, the abstract/article was assessed by a third reviewer and inclusion determined by consensus. Further, if an abstract/article was on the reference list of a retained article and its title contained ‘disintegrative disorder’, ‘regressive autism’, ‘autistic regression’ or ‘developmental regression’, it was included if it satisfied the eligibility criteria. Thus, articles before 2010 were included if they had impacted our area of interest.
Strength of Evidence
We developed an objective, 8-tiered tool to assess the strength of evidence (Table 1). Because of the nature and heterogeneity of included studies (see Study Properties section), neither a risk of bias nor a meta-analysis was conducted.
Assessment of the Strength of Evidence of Included Papers.a

Reviews and non-research papers were not scored.
The assessed strength of evidence is relative only to that of other papers in the review.
Categories
Papers were categorized as genetic associations, immune associations, and/or comorbid associations.
Results
Search Results
From database searches, 73 papers were retrieved. Twenty-four remained after the exclusion of 49 papers due to (1) being a duplicate of a previous inclusion, (2) English full text being unavailable or (3) not relating to the research question. From our search of reference lists of included papers, 8 more papers were added, making a total of 32 papers (Figure 1).
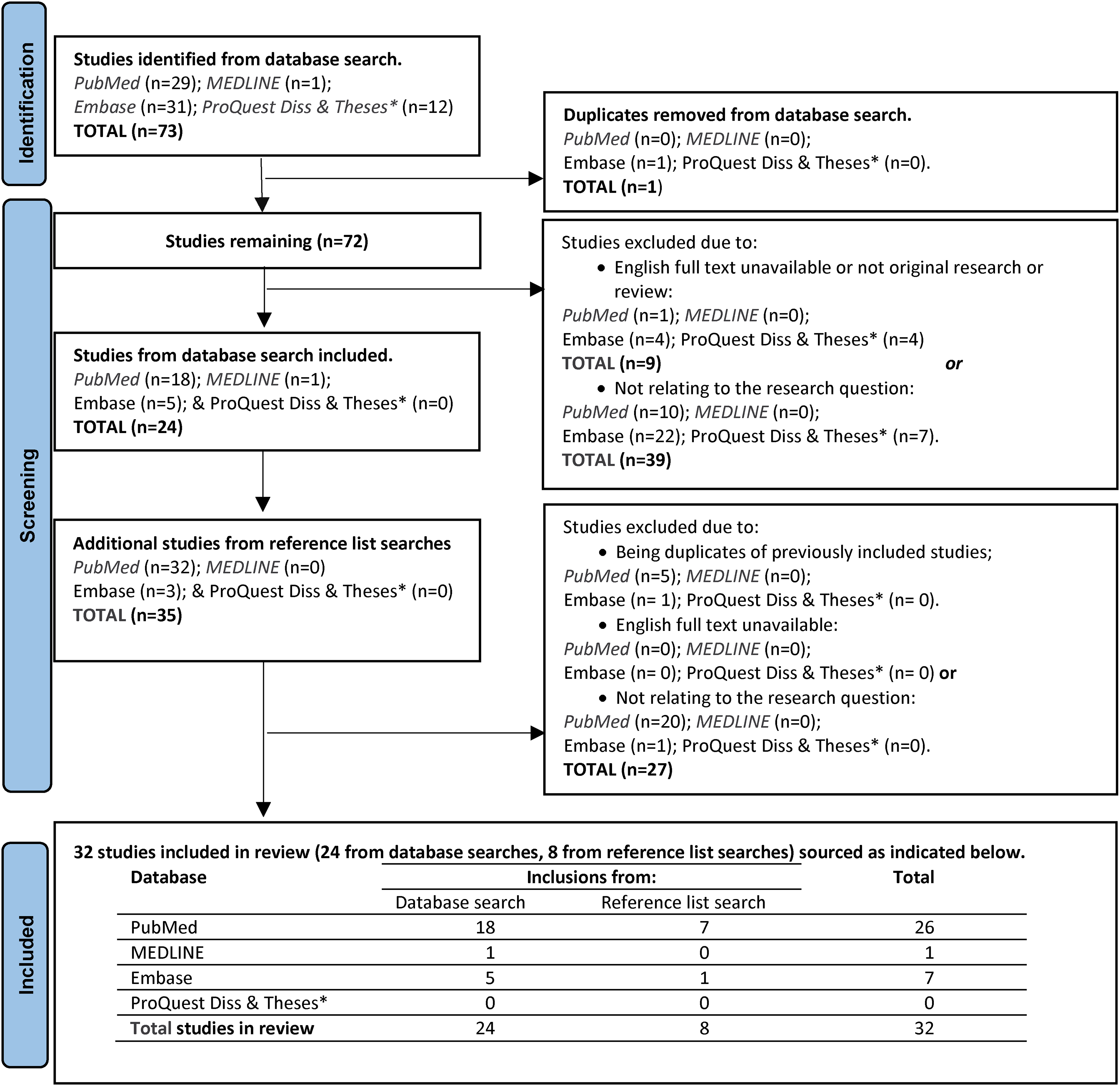
Identification of included studies. Note: Proforma for flowchart from PRISMA (https://www.prisma-statement.org/prisma-2020-flow-diagram).
Study Properties
Properties of papers are summarized in Table 2. Of the included papers, 11 (34%) originated in the United States; 4 (12%) in Italy; 3 (9%) in each of India, France, and China; 2 (6%) in Canada and 1 (3%) in each of Israel, Sweden, Morocco, Netherlands, Japan and the United Kingdom. Of these, 13 were case studies; 3, observational studies; 9, case-control (C-C) studies; 2, animal studies; 2, case series studies; 2, reviews; and 1, a multidisciplinary study. In 20 papers, RA or broader RA was a primary interest, CDD in 9, and Down syndrome disintegrative disorder (DSDD) in 3.
Properties of Studies in the Review.a
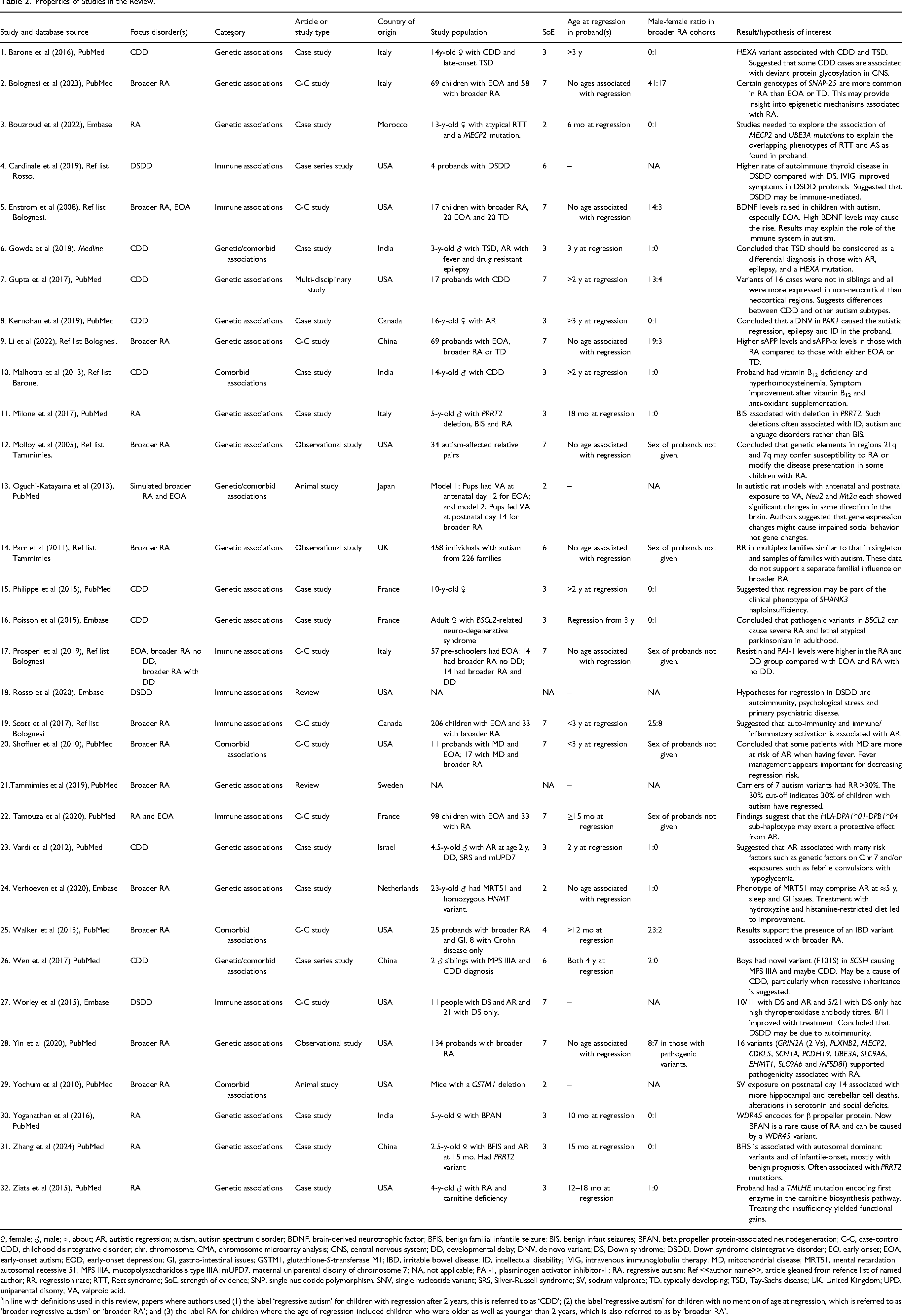
♀, female; ♂, male; ≈, about; AR, autistic regression; autism, autism spectrum disorder; BDNF, brain-derived neurotrophic factor; BFIS, benign familial infantile seizure; BIS, benign infant seizures; BPAN, beta propeller protein-associated neurodegeneration; C-C, case-control; CDD, childhood disintegrative disorder; chr, chromosome; CMA, chromosome microarray analysis; CNS, central nervous system; DD, developmental delay; DNV, de novo variant; DS, Down syndrome; DSDD, Down syndrome disintegrative disorder; EO, early onset; EOA, early-onset autism; EOD, early-onset depression; GI, gastro-intestinal issues; GSTM1, glutathione-S-transferase M1; IBD, irritable bowel disease; ID, intellectual disability; IVIG, intravenous immunoglobulin therapy; MD, mitochondrial disease; MRT51, mental retardation autosomal recessive 51; MPS IIIA, mucopolysaccharidosis type IIIA; mUPD7, maternal uniparental disomy of chromosome 7; NA, not applicable; PAI-1, plasminogen activator inhibitor-1; RA, regressive autism; Ref <<author name>>, article gleaned from refence list of named author; RR, regression rate; RTT, Rett syndrome; SoE, strength of evidence; SNP, single nucleotide polymorphism; SNV, single nucleotide variant; SRS, Silver-Russell syndrome; SV, sodium valproate; TD, typically developing; TSD, Tay-Sachs disease; UK, United Kingdom; UPD, uniparental disomy; VA, valproic acid.
In line with definitions used in this review, papers where authors used (1) the label ‘regressive autism’ for children with regression after 2 years, this is referred to as ‘CDD’; (2) the label ‘regressive autism’ for children with no mention of age at regression, which is referred to as ‘broader regressive autism’ or ‘broader RA’; and (3) the label RA for children where the age of regression included children who were older as well as younger than 2 years, which is also referred to as by ‘broader RA’.
Papers were placed into at least 1 category, using the prominent area(s) of their investigations into RA or CDD. There were 21 in Genetic Associations; 7 in Immune Associations; and 7 in Comorbid Associations. Three papers were in 2 categories (Table 2).
Genetic Associations
Papers are described within the categories of Heritability of Regression; Autosomal Variants; Chromosomal Variants; Sex-Linked Variants; More Complex Inheritance; Pathogenicity; and Epigenetic Associations, Toxicant Exposures and Gene Expression while referencing the Strength of Evidence descriptors (Table 1). This is followed by a summary and a tabulation (Table 3) of pertinent information gleaned from the papers.
Properties of Gene Variants Associated with Regression.
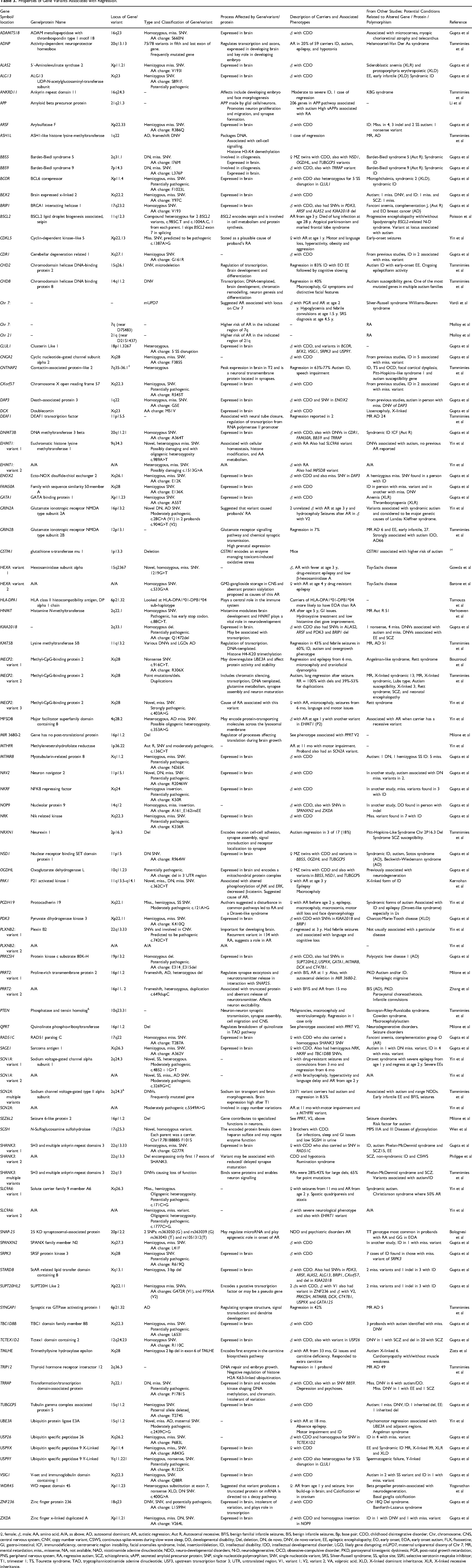
♀, female; ♂, male; AA, amino acid; A/A, as above; AD, autosomal dominant; AR, autistic regression; Aut R, Autosomal recessive; BFIS, benign familial infantile seizures; BIS, benign infantile seizures; Bp; base pair; CDD, childhood disintegrative disorder; Chr, chromosome; CNS, central nervous system; CNV, copy number variant; CSWS; continuous spike-waves during slow wave sleep; DD, developmental disability; Del, deletion; DN, de novo; DNV, de novo variant; EE, epileptic encephalopathy; EO, early onset; EOA, early onset autism; FLX, fluoxetine; GI, gastro-intestinal; ICF, immunodeficiency, centromeric region instability, facial anomalies syndrome; Indel, insertion/deletion; ID, intellectual disability; IDD, intellectual developmental disorder; LGD, likely gene disrupting; mUPD7, maternal uniparental disomy of Chr 7; MR, mental retardation; miss., missense; NA, not available; NAD, nicotinamide adenine dinucleotide; NDD, neuro-developmental disorders; N-D, neurodegenerative; OCD, obsessive-compulsive disorder; PKD, paroxysmal kinesigenic dyskinesia; PGR, post-natal growth retardation; PNS, peripheral nervous system; RA, regressive autism; SCZ, schizophrenia; sAPP, secreted amyloid precursor protein; SNP, single nucleotide polymorphism; SNV, single nucleotide variant; SRS, Silver-Russell syndrome; SS, splice site; SSRI, selective serotonin reuptake inhibitor; T1, trimester 1; TS, Tourette syndrome; TAD, tryptophannicotinamide adenine dinucleotide; USF3, upstream transcription factor 3; UTR, untranslated region; V1, variant 1; V2, variant 2; VA, valproic acid; XLD, X-linked dominant inheritance; XLR, X-linked recessive inheritance.
Heritability of Regression
Results are conflicting. In a review, 4 2 studies reported only a moderate heritability of regression. Of these, one 15 included 71 monozygotic (MZ) and 126 dizygotic (DZ) twin pairs and heritability was assessed using 3 intra-class correlations for language regression, social skills and broader regression within each twin group. In the MZ group all 3 correlations were low (around 0.3). Correlations for the DZ group were very low (0.019 or less). A higher level of evidence was provided by others 16 using data from 179 sibling pairs with autism. The concordance of autism and autistic regression was only 19%, not significantly higher than that expected with independence. Authors concluded that their results did not support separate genetic influences on autism and regression. 16 Contrastingly, the review 4 also reported 2 results of a study 17 providing stronger evidence of the concordance of autism and autistic regression. First, of 27 DZ twins with autism, the concordance of autism and loss of any skills was 67%. Second, of 25 MZ twin pairs with autism, only 2 were discordant for regression. 17 These disparate results suggest that environment is playing a role as well as genetics.
Autosomal variants
Higher-level evidence associated with CDD was provided by a multi-disciplinary study. 7 Using whole exome sequencing of 17 probands with CDD, 1 or more rare gene variants, not present in typically developing (TD) siblings, were found in 16. Along with 3 other autosomal variants, a de novo (DN), missense single-nucleotide variant (SNV) of BBS5 was found in female MZ twins and a BBS9 SNV in a male proband carrying another autosomal variant. Syndromic intellectual disability (ID) had previously been associated with variants of BBS. 7
Weaker evidence of variants associated with CDD was found in a case study. 18 Six former patients, each afflicted with a fatal neurodegenerative disease of infant onset, were compared to the female proband with CDD. All 6 of the former patients were homozygous or compound heterozygous for a BSCL2 variant and most died of pulmonary infection at 7 or 8 years. The female proband regressed after 3 years and, as an adult, developed atypical Parkinson disease. She also died of pulmonary infection. Her relevant genotype was likely compound heterozygosity for 2 variants affecting the splicing of exon 7 in BSCL2 18 (Table 3).
Researchers 19 provided higher-level evidence pertaining to broader RA. To start, a literature search identified 47 genes associated with broader RA. These genes were then sequenced in a cohort of 134 individuals with broader RA to identify variants in the group. Two unrelated male probands each had the same DN SNV in GRIN2A and a third male carried another GRIN2A variant. These variants, occurring in 3 of a cohort of 134, prompted the authors 19 to propose that they might be causal of autistic regression in these males. Also in the cohort was a female proband with a novel, missense SNV in UBE3A. Her regression began at 18 months and associated symptoms were epilepsy, ID, quadriparesis and balance issues. Another was a female with a missense, hemizygous SNV in SLC9A6. She had seizures from 11 months and autistic regression at 2 years. Another 2 males each carried a variant in the same gene. The first carried a novel, heterozygous missense SNV in EHMT1, along with an X-linked SNV. In the second, heterozygous missense variants were identified in EHMT1 and MFSD8. At 8 years, clinicians noted the boy's severe cognitive and language delay and proposed that his regression may have been due to a partial loss of function in the pathways of EHMT1 and MFSD8. Persons with autism have previously been found to carry DN variants (DNVs) in GRIN2A and EHMT1. Variants in EHMT1 have not been associated with broader RA before and only homozygous variants in MFSD8 have been previously associated with broader RA 19 (Table 3).
Two case studies20,21 provided preliminary evidence connecting HEXA variants to broader RA. Each proband had Tay Sachs Disease (TSD). In the first, 20 a girl with a homozygous SNV in HEXA, had CDD and refractory epilepsy from 3 years. In the second, 21 a boy with a homozygous but different SNV in HEXA had CDD and refractory epilepsy from 4 years (Table 3). Another case study 22 described 7 members of 2 consanguineous, but unrelated, families. In each, 1 member had a different HNMT missense SNV, along with severe ID, speech delay, and regression after 5 years. The condition was named ‘mental retardation, autosomal recessive 51’ (MRT51). The study focus was a male with MRT51 and homozygous for an HNMT variant. He exhibited aggression, speech delay, autistic traits and gastro-intestinal (GI) issues. Treatment with hydroxyzine, antihistamine and a histamine-restricted diet led to sustained and significant improvement 22 (Table 3).
Preliminary evidence of 2 case studies23,24 linked a PRRT2 variant and RA. The first 23 described a boy with a heterozygous PRRT2I deletion and an autosomal deletion. He had MRT51 with autistic regression at 1 year. The second 24 described a girl with benign familial infantile seizures [BIS (familial)] and a heterozygous, frameshift PRRT2 duplication. She developed typically until exhibiting autistic regression at 15 months. Her family had variable phenotypic expression of the PRRT2 mutation. Mother, brother and proband were all carriers, with the mother being asymptomatic, the brother having BIS (familial) and the proband, RA 24 (Table 3).
Chromosomal variants
Higher level evidence connected gene variants in particular chromosomal regions to broader RA. 25 Using data from the Autism Genetic Resource Exchange Database, researchers defined an affected relative pair (ARP) as 2 probands with autism who were first- or second-degree relatives. Linkage analysis investigated autism susceptibility loci pertaining to broader RA in 34 ARPs with regression, and 151 without regression. Results suggested that changes in genes on chromosomes 7 and 21 rendered regression more likely 25 (Table 3).
Further preliminary evidence associated chromosome 7 and RA. 26 The primary interest was a boy with post-natal growth retardation, 2 febrile convulsions, mild hypoglycemia and autistic regression at 18 months. A maternal uniparental disomy of chromosome 7 (mUPD7) was identified, and the boy was subsequently diagnosed with Silver-Russell syndrome. The authors 26 proposed that his RA was due to combined genetic and environmental risk factors such as an mUPD7 and hypoglycemic events (Table 3).
Sex-linked variants
An observational study 27 identified a male and female with the same SNV in PLXNB2. The male suffered febrile seizures and autistic regression at 36 months whereas the female regressed at 14 months. Authors suggested that this SNV played a role in these regressions. Two other probands carried different SCN1A variants. The first was a heterozygous, novel, splice site variant in a female who regressed from 6 months and with febrile seizures and convulsions from 3 months. The second was also a heterozygous, novel variant in a male. He presented with brachycephaly, language delay and autistic regression at 2 years. In the same study, 27 2 homozygous SNVs in SCN2A and MTHFR were located in a male proband with motor impairment and autistic regression at 11 months. 27 Others 4 reported that around a third of carriers of SCN2A variants had autism. Regression was documented in 8.5% of cases (Table 3).
Deletions and point mutations in SHANK3 have been implicated in RA with a research group 7 reporting a homozygous, missense variant in SHANK3 in a female with CDD. Further, but lower-level evidence of a variant associated with CDD and SHANK3 was found in a case study 28 focussing on a girl with CDD carrying an interstitial SHANK3 deletion. Authors considered that this variant was associated with her disorder. A review 4 associated SHANK3 with RA. Those with large deletions in SHANK3 had a 28% to 43% regression rate (RR), whereas those with point mutations had a 65% RR. Associated phenotypes of SHANK3 included autism, ID and schizophrenia 4 (Table 3).
A C-C study 12 provided higher level evidence of an association of a single nucleotide polymorphism (SNP) with broader RA. The frequency of 2 single SNPs in SNAP-25 was investigated in children with broader RA (n = 58) and EOA (n = 69). In the broader RA group, the rs363039 G allele and GG genotype were significantly less frequent than in the EOA group. On the other hand, in the broader RA group, the rs1051312 T allele and TT genotype were significantly more frequent than in the EOA group. The authors decided that these SNPs might be useful biomarkers for RA (Table 3).
Along with an autosomal variant, researchers 7 found that a male with CDD had a DN, missense TRRAP SNV, previously reported in a person with autism (Table 3). There were also reports of a DN, missense SNV in NAV2 in a male with CDD whilst another male with CDD had a homozygous insertion in NOP9, along with 2 X-linked variants. In a third male with CDD, 7 a DN SNV in ZNF236 was identified. Other studies were cited which reported autism in 2 carriers of missense SNVs in NAV2. 7 A fourth male with CDD, with 4 variants on the X and another on the Y chromosome, was heterozygous for an SNV in CLUL1. The same study reported that female MZ twins carried a DN deletion in the 3´UTR region of OGDHL which had previously been associated with neurodegeneration. These girls also carried a DN SNV in NSD1 and a hemizygous SNV in TUBGCP5. Both SNVs had been identified in probands with autism and ID before. 7 Apart from 6 hemizygous SNVs, a male with CDD had a homozygous deletion in PRKCSH. 7 Along with a second autosomal SNV, a female proband 7 carried a homozygous missense SNV in RAD51C. In addition to 3 hemizygous mutations, 2 homozygous deletions in KIAA2018 and a homozygous, missense SNV in BRIP1, were identified in a male with CDD. 7 Nonsense, missense DNVs in KIAA2018 have been found in individuals with autism before. 7 Along with a hemizygous variant, another male with CDD had a homozygous variant in TCTEX1D2 which had been previously associated with schizophrenia 7 (Table 3).
A male with CDD carried a homozygous SNV in DNMT3B, along with 2 DN, autosomal variants and 2 X-linked SNVs. This DNMT3B variant had formerly been reported in persons with syndromic ID. 7 Along with a hemizygous, missense SNV, a second male with CDD carried a homozygous, missense SNV in DAP3 whilst a homozygous, missense SNV in ADAMTS18 was identified in a third male with CDD 7 (Table 3).
Preliminary evidence associating a gene variant to CDD was provided by a case study 29 assessing a female proband with autistic regression from 3 years, epilepsy and macrocephaly. Genetic sequencing identified a heterozygous, DN, missense PAK1 SNV. Authors 29 suggested a link between CDD and the dominant PAK1 SNV. Similar preliminary evidence was provided by others 23 where SEZ6L2 and QPRT deletions were identified in a male with BIS and RA. Variants of SEZ6L2 have been reported in persons with autism and QPRT variants in persons with neurodegeneration 23 (Table 3).
A review 4 described variants associated with autistic regression. A DN microdeletion in CHD2 was associated with broader RA in 5 of 6 carriers. The resultant phenotype was early-onset epileptic encephalopathy followed by cognitive loss. A CHD8 variant had a 40% RR in carriers. Carrier traits included macrocephaly, GI symptoms and distinctive facial features. With an RR of 43%, 60% of carriers of heterozygous KMT5B variants had febrile seizures. Five of 8 (63%) carriers of biallelic CNTNAP2 variants whereas only 1 of 7 (14%) carriers of heterozygous variants in CNTNAP2 exhibited regression. Of 17 carriers with a deletion in NRXN1, only 3 (18%) exhibited regression. There was also reported regression in 2 probands with DEAF1 variants, 1 in a carrier of a PTEN variant and 1 in a carrier of a TRIP12 variant. The same review reported that from 41% (7 of 17) to 80% (4 of 5) of carriers of SYNGAP1 variants were in probands with RA. Contrastingly, ADNP variants were associated with regression in only 20% of 59 carriers. There was reported regression in a carrier of an ANKRD11 variant and a carrier of an ASH1L) DNV. Regression occurred in 2 carriers of GRIN2B variants and authors 4 considered that these GRIN2B variants were associated with autism (Table 3).
A case series study 30 provided weaker evidence associating an SGSH variant with CDD. Here, 2 brothers, homozygous for a novel variant in SGSH, experienced autistic regression from 4 years (Table 3).
Sex-linked variants
An observational study 27 identified a novel missense SNV in MECP2 in a 7-year-old girl with microcephaly, seizures from 6 months and subsequent RA. Her regression was attributed to the SNV but it was unclear as to whether her diagnosis was Rett syndrome (RTT). Preliminary evidence of a case study 31 described atypical RTT in a girl with a nonsense SNV in MECP2. She had epilepsy from 6 months, followed by impeded development, regression, microcephaly and craniofacial dysmorphia. A review 4 contained 2 studies reporting on MECP2 duplication syndrome. One 32 comprised 31 probands with RTT and of these, 12 (39%) experienced autistic regression after an initial seizure. The second 33 comprised 17 probands, mostly with autism. Language regression was reported in 8 (47%), and regression in other areas in 7 (41%). See Table 3.
Two males with CDD 7 carried 2 different hemizygous, missense SNVs in SUPT20HL2. One also had an autosomal, DN SNV and the other, an autosomal, homozygous deletion, and 5 hemizygous, missense, X-linked SNVs (Table 3).
Beta propeller protein–associated neurodegeneration (BPAN) is a single gene disorder involving neurodegeneration due to iron accumulation in the brain. A case study 34 provided preliminary evidence connecting WDR45 to RA. It featured a 5-year-old girl with autistic regression from 1 year, seizures, intracranial calcification and iron accumulation in the brain. Her diagnosis of BPAN was further supported by a heterozygous, nonsense, DN mutation in WDR45 which had previously been associated with BPAN. 34 Another case study 35 provided preliminary evidence. It focused on a 4-year-old boy with GI issues, autistic regression from 33 months and carrying a hemizygous, 2-basepair deletion in exon 6 of TMLHE. Testing indicated a carnitine deficiency, and with carnitine supplementation, his functioning improved (Table 3).
Missense SNVs in NRK, TBC1D8B, NKRF and SAGE1 were identified in a boy with CDD. 7 In addition to 2 autosomal variants, another male carried hemizygous, missense SNVs in PDK3, ARSF, ALAS2, Cxorf57 and ALG13, and a deletion in STARD8. Another male with CDD carried a missense SNV in MTMR8. Authors 7 reported that SNVs in NRK, NKRF, Cxorf57, ALG13 and STARD8 had been identified in people with ID, SNVs in TBC1D8B in individuals with autism and ID, and variants in SAGE1I, MTMR8 and ARSF in individuals with autism and individuals with ID. 7
The same study 7 also identified missense, hemizygous SNVs in CDR1 and FAM50A in a boy with CDD whilst another carried a hemizygous missense SNV in CNGA2. Along with an autosomal variant, another male with CDD carried a hemizygous missense SNV in USP26. In addition to an autosomal variant, another carried an X-linked missense SNV in ENOX2 whilst another, along with an autosomal variant, carried a hemizygous, missense, DN SNV in ZXDA and a hemizygous, missense SNV in SPANXN2. Variants in these genes have previously been associated with ID. 7 Another male with CDD was heterozygous for an autosomal mutation, carried hemizygous missense SNVs in BCOR, BEX2, SRPK3 and VSIG1, and a fifth on the Y-chromosome in USP9Y. Authors 7 reported that previous studies had found ID in probands with variants in BCOR, BEX2, SRPK3 and VSIG1, with autism also identified in 2 with a splice site variant in VSIG1 and in 1 with a hemizygous, missense SNV in BEX2 (Table 3).
A study 27 described a female with a missense SNV in CDKL5 and considered this a likely cause of her RA from 1 year. Another had an X-linked, missense SNV in PCDH19. Symptoms included autistic regression from 16 months, epilepsy and a Dravet-like syndrome. Authors proposed a disruption of shared pathways of RA and the Dravet-like syndrome had led to her regression 27 (Table 3).
More complex Inheritance
A C-C study 6 provided higher level evidence linking genetics to broader RA. Authors wrote that more than 200 genes associated with autism were in the metabolic pathway of the amyloid precursor protein. Secreted amyloid precursor protein–a (sAPPa) is produced when amyloid precursor protein is cleaved by a secretase during non-amyloidogenic processing. This can result in amyloid plaques, found in the brains of some autistic children. Moreover, high levels of sAPPa can impede brain development during important periods. Plasma levels of sAPPa were significantly elevated in children with broader RA compared to those with EOA and TD children. Authors 6 suggested that higher sAPPa levels may be a useful biomarker for broader RA (Table 3).
A review 4 described a study, using family data, to estimate the involvement of likely gene-disrupting (LGD) DNVs in RA vs EOA. Authors focused on genes encoding for fragile X mental retardation protein, diseases of Mendelian inheritance, chromatin modification, postsynaptic density proteins and proteins expressed embryonically. The LGD DNVs encoding for postsynaptic density proteins were more frequent in the broader RA group but small numbers limited any conclusion. 4
Pathogenicity
Established methods 7 assessed variant pathogenicity in the 4 areas of brain expression, conservation, intolerance to functional variation and likelihood of being damaging. Of 40 candidate genes, 14 (NRK, TBC1D8B, TRRAP, NAV2, OGDHL, ZNF236, PRKCSH, MTMR8, BCOR, SRPK3, USP9Y, KIAA2018, CXorf57 and ALG13) met each criterion and were described as ‘potentially pathogenic’. Others 27 used the criteria of the American College of Medical Genetics and Genomics to assess variant pathogenicity. 36 Sixteen variants of 12 genes met 1 criterion of pathogenicity and were classified as variants of uncertain significance. Both GRIN2A variants, both SCN1A variants, along with SCN2A, MTHFR, PCDH19, UBE3A and EHMT1 (first variant described in Table 3) variants were classified as ‘moderately pathogenic’ (PM2). Both PLXNB2 variants and both SLC9A6 variants, along with CDKL5, MFSD8I and EHMT1 (second variant described in Table 3) variants, were classified as ‘supporting pathogenic’ (PP3), while the MECP2 variant was deemed ‘strongly pathogenic’ (PS1). See Table 3. Contrastingly, a third group 22 described an HNMT variant as pathogenic based on trio-based exome sequencing (Table 3).
Epigenetic associations, toxicant exposures, and gene expression
A C-C study 12 provided stronger evidence of epigenetics in relation to broader RA. An allele of SNAP-25, a target of microRNA, was significantly more common in probands with broader RA than in TD or EOA probands. The findings of an animal study 14 suggested that in combination, timed toxicant exposure after birth and a particular genotype resulted in social regression in mice. Researchers investigated whether mice with a GSTM1 deletion were more sensitive to sodium valproate exposure postnatally than wild-type mice. The genetically altered mice were injected with sodium valproate or saline on post-natal day 14. Mice exposed to sodium valproate had increased neuronal losses and over time, sodium valproate exposure resulted in changed brain chemistry and less sociability. 14
Lesser preliminary evidence was provided by a C-C study using autistic rat models. 37 Authors aimed to explore aberrant social behavior and anxiety in relation to broader RA and EOA after exposure to valproic acid. To emulate EOA, 5 pups of 5 dams were administered equal amounts of valproic acid on embryonic day 12 (Model 1). To emulate broader RA, 5 pups of 5 dams were administered equal amounts of valproic acid on post-natal day 14 (Model 2). Pups from each model exhibited aberrant sociability and anxiety. Microarray analysis compared the gene expression profiles of Model 1 and Model 2 pups. In both models, Neu2 and Mt2A changed expression in the same direction. Researchers concluded that with exposure to neurotoxins at different developmental stages, gene expression changes rather than gene changes may have a stronger effect on sociability and anxiety. 37 A case study 38 supplied preliminary evidence relating to exposure to neurotoxins and CDD. A 14-year-old vegetarian male patient had typical development until 6½ years. Thereon, he was fearful, with fading speech, echolalia and emergent autistic behaviors and an eventual diagnosis of CDD. Testing showed low serum vitamin B12, raised plasma homocysteine levels, and panhypoperfusion of the brain. After vitamin B12, anti-oxidant and other supplementations, improvement was noted. Clinicians reported that reduced vitamin B12 levels may have caused the neurotoxicity and regression. 38
A study 7 found that their 40 candidate genes for CDD were more highly expressed in non-neocortical regions than neocortical regions and that their expression profile was similar to that of an independent cohort with autistic regression. Further, expression was increased during the developmental periods of 1-6 years and 6-11 years, which is when probands’ symptoms emerged. They then compared the median expression levels of non-synonymous and synonymous variants in non-neocortical and neocortical regions in probands from the 4 groups of CDD, TD siblings, RA and EOA. With 1 exception, the expression of the candidate genes for CDD was distinctive and qualitatively different from the profiles of other gene sets. The exception was in the non-synonymous variants of probands with RA. This was despite probands with CDD and probands with RA having only a NAV2 variant in common. Extending the analysis to other genes sets showed that the gene expression profile of non-synonymous variants in probands with CDD and probands with RA were both distinctive and qualitatively different from the other gene set profiles. 7
A C-C study 39 provided low-level evidence regarding gene expression and broader RA. Associations between broader RA and GI issues were explored in 4 groups. A case group comprised 25 children with broader RA from 12 months and comorbid GI issues. Three control groups were children with: Crohn disease, no autism; Ulcerative colitis, no autism; and Neither inflammatory bowel disease nor autism. For all children, transcriptome profiling of GI mucosal biopsy tissue–enabled gene expression for each group to be quantified. Results suggested a unique gene expression profile associated with inflammatory bowel disease in children with broader RA and GI issues. 39
Summary
The reported concordance rates of autism and broader RA were inconsistent. In broader RA, 86 gene variants, 3 chromosomal regions, and 1 metabolic pathway were described. Of the genes, 33 were X-linked, 1 Y-linked, and 52 autosomal. Apart from the X-chromosome, the most common variant locations were chromosome 16 (7) and chromosomes 2 and 11 (6 each). Only 10 variants were novel. Hemizygous (vs homozygous or heterozygous) was the most stated variant zygosity. Less common references to zygosity were oligogenic heterozygosity, for 4 variants, and compound heterozygosity, for 1. At the DNA level, there were 57 SNVs, 15 deletions, 3 frameshift variants, 4 variants at splice sites, 3 duplications, 1 insertion, and 3 CNVs or associated with CNVs. In terms of protein production, there were 38 missense and 4 nonsense variants, an LGD variant and a loss of function variant. With respect to origin, 19 variants were DN and 2 were of maternal origin (Table 3).
Treatments improving the functioning of probands with broader RA were described in conjunction with variants of HNMT and TMLHE. Frequently associated phenotypes of broader RA were epilepsy and macrocephaly. Conditions most often related to variants in those with broader RA were ID, autism and schizophrenia. Most genes were cited only once, with exceptions being MECP2 and SHANK3, each cited in 3 studies, and EHMT1, HEXA, PLXNB2, PRRT2, SCN1A, SCN2A and SLC9A6, each cited in 2 (Table 3). Of the described variants, 58 were in probands with CDD and 37 in probands with broader RA but not necessarily CDD.
Higher levels of sAPPa in children with broader RA may become a useful biomarker. 6 Two studies7,21 assessed the pathogenicity of variants associated with broader RA using established criteria. Of variants assessed, about a third met 1 of more of the criteria for pathogenicity.
Differential frequency of 2 SNAP-25 alleles in probands with broader RA from those with EOA suggested epigenetic involvement in broader RA. 12 A case study 38 and 2 animal C-C studies14,37 associated toxicant exposure to broader RA. In the case study, a boy with low serum vitamin B12 and raised plasma homocysteine levels was diagnosed with CDD. In the first animal study, rats exposed to valproic acid postnatally developed impaired sociability, and in the second, mice with a GSTM1 deletion exposed to sodium valproate postnatally developed impaired sociability.
Candidate genes for CDD were differentially expressed in non-neocortical regions from neocortical regions, and non-synonymous variants of these genes were expressed in a similar way to those of probands with RA but not probands with EOA. 7 Unique gene expression was identified in children with broader RA and GI issues compared with other groups. 39
Immune Associations
Associations of autistic regression and the immune system are described in the categories of broader regressive autism and regression in Down syndrome, followed by a summary.
Broader regressive autism
Higher level evidence of differences in brain chemistry pertaining to the immune system in those with broader RA, compared to those with EOA, were reported by a C-C study. 40 Proinflammatory cytokines, plasminogen activator inhibitor-1 (PAI-1), and resistin levels were compared in children with broader RA, with and without developmental disability and children with EOA. The mean level of proinflammatory cytokines did not differ between the combined broader RA and EOA group. However, resistin and PAI-1 levels were higher in the broader RA with DD group than in the other 2 groups. 40
The BDNF gene encodes brain-derived neurotrophic factor (BDNF) produced by activated brain microglial cells. These cells share features with peripheral macrophages, suggesting that BDNF production has an important role in the immune system. A C-C study 41 produced higher level evidence of a connection between the immune system and broader RA. Among the groups of neonates compared were those who developed broader RA and those who were later diagnosed with EOA. Comparing blood samples of the broader RA and EOA groups found increased BDNF levels (though not significantly) in the EOA group. Authors hypothesized that BDNF levels may be associated with autism development and altered for those with broader RA. 41 Other studies42,43 provided higher level evidence of an immune connection with broader RA. In the first, 43 medical charts of children with autism were reviewed. Autoimmune conditions in first- and second-degree relatives were recorded according to whether the child had broader RA (n = 33) or EOA (n = 206). Autoimmune thyroid disease and familial type 1 diabetes were significantly more common in broader RA families, suggesting that those with broader RA were more prone to immune issues than those with EOA. 43 In the second, 42 haplotypes of the human leukocyte antigen (HLA) system, in 33 children with broader RA and 98 with EOA, were examined. Of those with broader RA, 62 (43%) had HLA-DPA1*01-DPB1*04 (DPA1) vs 14 (63%) with EOA, suggesting that DPA1 offers some protection from regression.
Regression in Down syndrome
Autistic regression can occur in people with Down syndrome and mostly during adolescence or early adulthood. 44 Down syndrome disintegrative disorder is one label for the resultant condition. A review 45 reported that most of those with DSDD have language regression and sometimes depression, social withdrawal, anxiety, psychoses and newly acquired insomnia. One study in the review investigated 4 patients with DSDD where treatment with benzodiazepines and electro-convulsive therapy resulted in recovery to baseline. 45
Mild evidence of autoimmune associations in people with DSDD came from a case series study 44 of 4 probands with DSDD, each carrying various auto-antibodies. After intravenous immunoglobulin treatment, all improved significantly, implying that regression in DSDD may be mediated by autoimmunity. 44 A C-C study 46 provided higher level evidence of a relationship between thyroid autoimmunity and DSDD. Compared with age-matched controls, 10 of 11 probands had higher concentrations of thyroperoxidase antibodies.
Summary
First, compared to probands with EOA and broader RA without DD, probands with broader RA and DD had higher resistin and PAI-1 levels. Second, compared to probands with EOA, probands with broader RA had lower BDNF levels as neonates and different allelic ratios in an HLA polymorphism than those with EOA. Third, there were higher proportions of autoimmune thyroid disease and familial type 1 diabetes in families of those with broader RA compared to those with EOA. Lastly, those with DSDD were more prone to autoimmune disorders than those with only Down syndrome.
Comorbid Associations
Inflammatory bowel disease and fever have each been associated with autistic regression. Low-level evidence of a comorbidity of broader RA and inflammatory bowel disease came from a C-C study 39 where results supported the existence of an inflammatory bowel disease variant in children with broader RA and GI issues. A C-C study 47 provided higher level evidence associating fever with autistic regression in persons with mitochondrial disease (MD). In 28 probands with autism and MD, autistic regression occurred in 17 (61%), significantly higher than the 30% in the broader autism population. Of those with broader RA, 12 (71%) regressed with fever. These data suggest that people with MD have an increased risk of broader RA and particularly with fever. 47 Autistic regression with fever was also described in a case study where the proband, who had TSD, regressed at 3 years with fever. 21
Preliminary evidence from a case study associated CDD with TSD and a HEXA variant 20 where the focus was a girl with late-onset TSD and an SNV in HEXA. She had suffered autistic regression from 4 years. Another case study 21 described a male proband with a different SNV in HEXA, who suffered autistic regression from 3 years. Testing showed that he was deficient in beta hexosaminidase A, prompting a diagnosis of TSD. A third case study 26 associated autistic regression at 2 years with Silver-Russell syndrome in a child with a mUPD7. Lesser preliminary evidence came from a case series study 30 which identified an association between mucopolysaccharidosis IIIA and CDD. In this study, 2 brothers with the same novel homozygous variant of SGSH regressed at 4 years. Authors hypothesized that the variant was the cause of their mucopolysaccharidosis IIIA and regression. 30
Discussion
A multitude of genetic variants were associated with broader RA7,27 and these support the heterogeneity of its genetic etiologies and correlates. A study of probands with broader RA suggested epigenetic involvement 12 whereas 3 studies14,37,38 associated toxicant exposures to broader RA. Some studies described differential expression of various genes and proteins in the brains 7 and GI mucosal tissue 39 of children with broader RA compared to controls. Other studies suggested immune involvement40–46 and future biomarkers for broader RA.6,12,39,40,42 Treatments improving proband functioning provide more clues as to the causes of broader RA22,35,38 and DSDD.45,46
Consistent Themes
Consistent findings supported the development of endotypes (also known as endophenotypes) from subtypes of broader RA. Endotypes consist of 1 or more clearly defined traits with a likely advantage of enabling the subtype to move from a phenotype closer to the underlying genetics. A possible endotype for broader RA is macrocephaly with comorbid seizure. These traits were coupled with broader RA in children with PCDH19 and PAK1 variants.27,29
With further research, carrying a particular variant(s), along with broader RA and macrocephaly with comorbid seizure might prove to be an endotype of broader RA, and 1 or more of the variants might become a biomarker for CDD, RA or broader RA. Along similar lines, various research papers reported that variants in ALG13, 7 CDKL5, 27 GRIN2A, 27 HEXA,20,21 HNMT,22,27 MECP2,27,31 PAK1, 29 PLXNB2, 27 QPRT, 23 SCN1A, 27 SEZ6L2, 23 SGSH, 30 SLC9A6, 27 TCTEX1D2, 7 TMLHE, 35 UBE3A, 27 USP26 7 and WDR45 34 were identified in probands with broader RA, along with GI issues,4,27,29,30,35 macrocephaly,4,27,29 and/or seizure.4,7,20,21,29,31,34
The male-female ratio in broader RA is close to the estimated 3:1 for autism. 48 The sex ratio of probands in this review at 151:51 (or 2.96:1) was consistent with this figure.
Researchers 25 identified a region of chromosome 21 associated with a higher risk of regression. In addition, adolescents with Down syndrome (or Trisomy 21) and autism are reported to have a risk of up to 50% of having regressed. 46 Increased dosage of key genes, in this region of chromosome 21, in adolescents with Down syndrome who develop DSDD, may contribute to regression by disrupting synaptic function, neuroinflammation, or mitochondrial activity.
Inconsistencies
Two studies from a review 4 provided disparate evidence of the heritability of regression. This may have been due to different definitions of regression and different methods to measure heritability. For example, in the first, 15 the age of regression was not stated and intra-class correlation coefficients were used to compare heritability for various types of regression, such as language, social and general regression, in MZ and DZ twins. In contrast, in the second study, 16 regression was prior to 3 years and described as early developmental regression and data were analysed using ARPs. One or more of these differences may have caused the conflicting results.
Inconsistencies emerged from comparisons of included papers with other papers. Four40–43 provided evidence that immune dysfunction plays a role in broader RA. Three papers44–46 provided similar evidence for DSDD. Low male-female sex ratios are characteristic of many immune disorders such as Sjogren syndrome (1:9) and rheumatoid arthritis (1:3) and with an overall ratio of 1:4. 49 Similarly, there is a low sex ratio in people with DSDD. For example, in 1 cohort, 46 the ratio of males to females was 4:7. On the other hand, the sex ratios in cohorts with broader RA are not reflective of immune dysfunction. For example, one study 41 reported 94% males in 17 individuals with broader RA and another 89% in 33 individuals with broader RA. 43
A study 40 compared groups of preschoolers with and without broader RA. The group with broader RA was further subdivided into those with DD and those without. This is inconsistent because autistic regression necessarily results in DD.
A research group 30 claimed that they had identified the genetic cause of CDD. However, CDD has many genetic causes not just one.
Future Research
A study exploring BIS 24 identified possible epigenetic or pleiotropic associations when 3 members of one family had the same gene variant but 3 distinct phenotypes, 1 of which was RA. With this in mind, and in the light of the rapidly increasing autism rates, it would seem important to focus on research elucidating how particular epigenetic or pleiotropic processes might increase the risk of broader RA.
A research group 27 wrote of 2 males with broader RA, one with missense variants in EHMT1 and MFSD8 and another with a different EHMT1 missense variant and another missense variant in SCL9A6. 27 They hypothesized that the regressions may have resulted from oligogenic events causing a partial loss of function in the pathways of the affected genes. 27 To our knowledge, oligogenic heterozygosity, involving small numbers of variants from multiple genes, has not been explored as a cause of broader RA. In this review of studies exploring variants,7,19 a major limitation to exploring oligogenic heterozygosity was the small cohort sizes (n = 17 and n = 134, respectively). Whole genome sequencing, aiming to explore oligogenic heterozygosity, with larger cohorts could further findings in the area.
Down the track, newly developed gene therapies may transform the treatment of broader RA with a known genetic cause. For example, recent research 50 described how CRISPR-Cas9 technology has the potential to transform the treatment of RTT by blocking the expression of the causal variant in patient cells.
Limitations and Strengths
A limitation of this review arises from the rarity of CDD and the omission of RA and CDD as diagnostic categories in DSM-5, likely resulting in fewer diagnoses, a lower perceived prevalence, less funding, and less related research. Hence, many of our included papers are case studies,18,20–24,26,28,29,31,34,35,38 case series studies,30,44 or other studies with relatively small cohorts 7 and thus providing weaker evidence than studies with larger cohorts.
The non-inclusion of RA and CDD from DSM-5 has also led to difficulties for researchers in consistently defining these disorders. This is important as differences between CDD, RA, and EOA can be underplayed or even lost if definitions vary. Some studies6,12,40–42 comparing children with RA and EOA, grouped all children with who had suffered autistic regression together, regardless of the age at regression. Moreover, 2 studies43,47 defined children with RA as having suffered regression before 3 years of age which overlaps with the definition of CDD. Thus, any results applying to just RA or just CDD would have been lost. In 8 articles,6,12,16,22,25,27,40,41 there was no age mentioned in association with regression. In 2 papers, children were included in the study population if they had reached 12 39 or 15 months 42 before regression. Again, different results for RA and CDD may have been missed. Possibly because of the exclusion of CDD from DSM-5 and the continued omission of RA as a diagnosis in any version of DSM, the definition of autistic regression and the criteria for recruiting affected children vary. Two studies4,19 described autistic regression as a loss of learned developmental skills such as language, motor or social skills whilst a third 40 defined regression as regression of skills. In each, there was no criterion relating to development prior to regression or relating to age at the commencement of regression. A different approach was taken in another study 35 where regressive groups were subdivided according to whether there had been developmental delays prior to regression.
A way forward to further explore the genetic and other etiologies of RA and CDD is made clearer by this review. Moreover, because about 30% of children with autism have broader RA 5 it follows that the more than 7-fold increase in autism prevalence this century 3 is echoed by a similar increase in broader RA. Hence, research into the causes and correlates of broader RA is becoming increasingly important. Furthermore, this review provides a comprehensive, ordered list of gene variants implicated in broader RA for clinicians, parents and researchers to explore in children with broader RA. Suggestions for researchers of areas to investigate in their search for answers are also provided.
Another strength of this review is the summary of recent studies pertaining to genetic etiologies and associations of broader RA that has not been done before. Further, we provide descriptions of relevant phenotypes and successful treatments. Importantly, conditions such as BIS (familial) 24 and fever in children with MD, 47 occasionally leading to broader RA, are described. This may result in more careful monitoring of vulnerable children, improved knowledge of autistic regression, and its prevention.
Our objective was to identify and summarize genetic and other associations of RA and CDD. This review accomplishes this by including and describing dozens of genetic associations, immune associations, and various comorbidities of RA and CDD.
Conclusion
Our review showed the heterogeneity of genetic etiologies and associations of broader RA. Potential biomarkers were suggested. Probands with broader RA had differential expression of various genes and proteins in the brain compared with controls. Possible immune, epigenetic or pleiotropic involvement, along with treatments with some successes, were also described.
Without exception, researchers used the definition for CDD provided by DSM-IV where regression is at 2 years or later. However, a range of definitions were used for RA. The limitations imposed by these varying definitions for RA would be reduced if researchers agreed on a standardised definition and used this in their research. In conjunction with a leading international autism association, a Delphi Panel could determine such a definition. One possibility is that the criteria for CDD used in DSM-IV 9 or International Classification of Diseases, 11th Revision (ICD-11) 51 is used to provide complementary criteria for RA regarding the age of regression. With the achievement of a standardised definition, research in this area would likely be more prolific and more able to be pooled enabling causes, prevention and treatments for broader RA to follow.
Because of the genetic heterogeneity of broader RA, it would be productive to explore 1 or more endotypes with the aim of future research being within an endotype. With research focused on exploring potential biomarkers within endotypes, clinicians would have heightened awareness of at-risk children and thus be more able to monitor their development. This might further our knowledge of broader RA and lead to improved outcomes for affected children.
As the prevalence of autism increases, so does the prevalence of broader RA. Around a third of children with autism have suffered regression, 5 and the estimated prevalence of autism in 2020 was 3.22%. 3 It follows that the estimated combined prevalence of broader RA is between 0.9% and 1%. There is no internationally agreed definition of a rare disease. However, the average prevalence range most commonly used is 40 to 50 cases per 100 000 (or 0.04 to 0.05%) people. 52 Hence, broader RA is no longer a rare disease. Research can lead to improved diagnostics, prevention and treatments, thus supporting patients and carers, improving their quality of life and reducing the fiscal burden on governments.
Footnotes
Author Contributions
JF conceived and designed this systematic review and performed the database search. All authors screened abstracts. JF wrote the original draft and FR, AG and JV provided critical appraisal and advice for JF which were used to produce subsequent drafts. All authors approved the final draft before submission.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.