
Abstract

Keywords
X-linked lymphoproliferative disease type 1 (XLP-1) is a rare primary immunodeficiency caused by pathogenic variants in the SH2D1A gene, which encodes signaling lymphocytic activation molecule–associated protein (SAP). 1 SAP is essential for T-cell, NK-cell, and invariant NKT-cell function; its deficiency results in profound immune dysregulation and a high risk of hemophagocytic lymphohistiocytosis (HLH), often triggered by Epstein-Barr virus infection. 1 CNS involvement in HLH is associated with significant morbidity and mortality.4 Early recognition and rapid initiation of HLH-directed therapy, followed by definitive hematopoietic stem cell transplantation (HSCT), can substantially alter neurologic and overall outcomes.2,3
Case Presentation
A three-year-old male presented with acute onset fever, progressive ataxia, and new-onset seizures. Neurologic examination demonstrated gait instability and altered mental status. Brain MRI obtained at presentation demonstrated extensive cerebral edema and multifocal T2-weighted / fluid-attenuated inversion recovery (FLAIR) hyperintensities involving the subcortical, deep, and periventricular white matter. Initial differential diagnosis included autoimmune encephalitis and infectious, neoplastic, or metabolic causes. However, laboratory findings of hyperferritinemia (149 ng/mL, normal 10-140 ng/mL), hypofibrinogenemia (128 mg/dL, normal 212-466 mg/dL), pancytopenia (white blood cell count 3300/µL, hemoglobin 9.8 g/dL, platelets 247 000/µL), and elevated IL2 receptor (21 925 pg/mL, normal 532-1891 pg/mL) met diagnostic criteria for HLH. 4 Genetic testing revealed a mutation in SH2D1A consistent with a diagnosis of XLP-1–associated HLH. Viral testing for Epstein-Barr virus was negative, and the patient was promptly initiated on CNS-directed HLH therapy.
Imaging Findings
Initial MRI showed diffuse bilateral white matter T2/FLAIR hyperintensities with associated cerebral edema. Diffusion-weighted imaging (DWI) and apparent diffusion coefficient (ADC) sequences demonstrated areas of restricted diffusion corresponding to regions of active inflammation, consistent with reported imaging patterns in pediatric CNS HLH. 5
Serial imaging during the acute phase demonstrated evolution of white matter changes and progression to parenchymal volume loss, reflecting ongoing inflammatory-mediated CNS injury. 5 Follow-up neuroimaging 2 years after escalation of therapy and transplantation showed stabilization of volume loss and partial improvement of signal abnormalities without further progression of cerebral atrophy. (Figure 1)
Therapeutic Intervention
Within 2 weeks from presentation, the patient was treated with HLH-directed therapy, including dexamethasone, etoposide, and intrathecal methotrexate, achieving initial stabilization. 4 Given refractory CNS disease and the underlying SH2D1A mutation, within 4 months from presentation, he underwent haploidentical bone marrow transplantation, the only curative therapy for XLP-1 associated HLH.2,3 Adjunctive immunomodulatory therapy with eculizumab and ruxolitinib was added to control immune activation. Post-transplant complications included mild graft-vs-host disease, managed with tacrolimus and mycophenolate mofetil, and adrenal insufficiency, managed with hydrocortisone.
Outcomes
Following bone marrow transplantation and immunomodulatory therapy, the patient showed significant neurologic improvement. Seizures were controlled on levetiracetam and lacosamide, with only mild residual left lower-extremity weakness. Serial MRIs at 2 years post treatment showed stabilization of parenchymal volume loss and partial resolution of white matter signal abnormalities, without further progression of cerebral atrophy. Functionally, the patient continued to improve with speech, language, and physical therapy, consistent with improved outcomes reported after early definitive therapy in XLP-1-associated HLH. 2
Discussion
CNS imaging findings in HLH are not pathognomonic and may overlap with other autoimmune, demyelinating, neurometabolic, or neoplastic entities. 5 Suspicion for CNS HLH was driven by the convergence of acute neurologic symptoms, fulfillment of systemic HLH criteria, and high-risk genetic association of XLP-1.1,4 CNS involvement is a major driver of morbidity and mortality in HLH, and a high index of suspicion is required in genetically at-risk patients. Bone marrow transplantation combined with targeted immunomodulatory therapy can stabilize and potentially reverse CNS manifestations in XLP-1–associated HLH. Prior cohort studies demonstrate significantly improved survival after transplantation, particularly when performed before irreversible organ damage occurs.2,3 This case supports a multidisciplinary, aggressive treatment approach in refractory CNS disease.
Conclusion
CNS HLH is a life-threatening complication of XLP-1. This case demonstrates that timely diagnosis, aggressive immunosuppression, and definitive treatment with bone marrow transplantation can result in meaningful neurologic recovery and radiologic stabilization. Early recognition and intervention remain critical to optimizing outcomes in this high-risk population.
2
Neuroimaging findings in XLP-1–associated HLH. (A-C) MRI at presentation demonstrates extensive, asymmetric bilateral supratentorial white matter involvement with facilitated diffusion consistent with vasogenic edema. (D-F) MRI 2 years post hematopoietic stem cell transplantation shows interval resolution of vasogenic edema and mild cerebral atrophy. HLH, hemophagocytic lymphohistiocytosis; MRI, magnetic resonance imaging; XLP-1, X-linked lymphoproliferative disease type 1.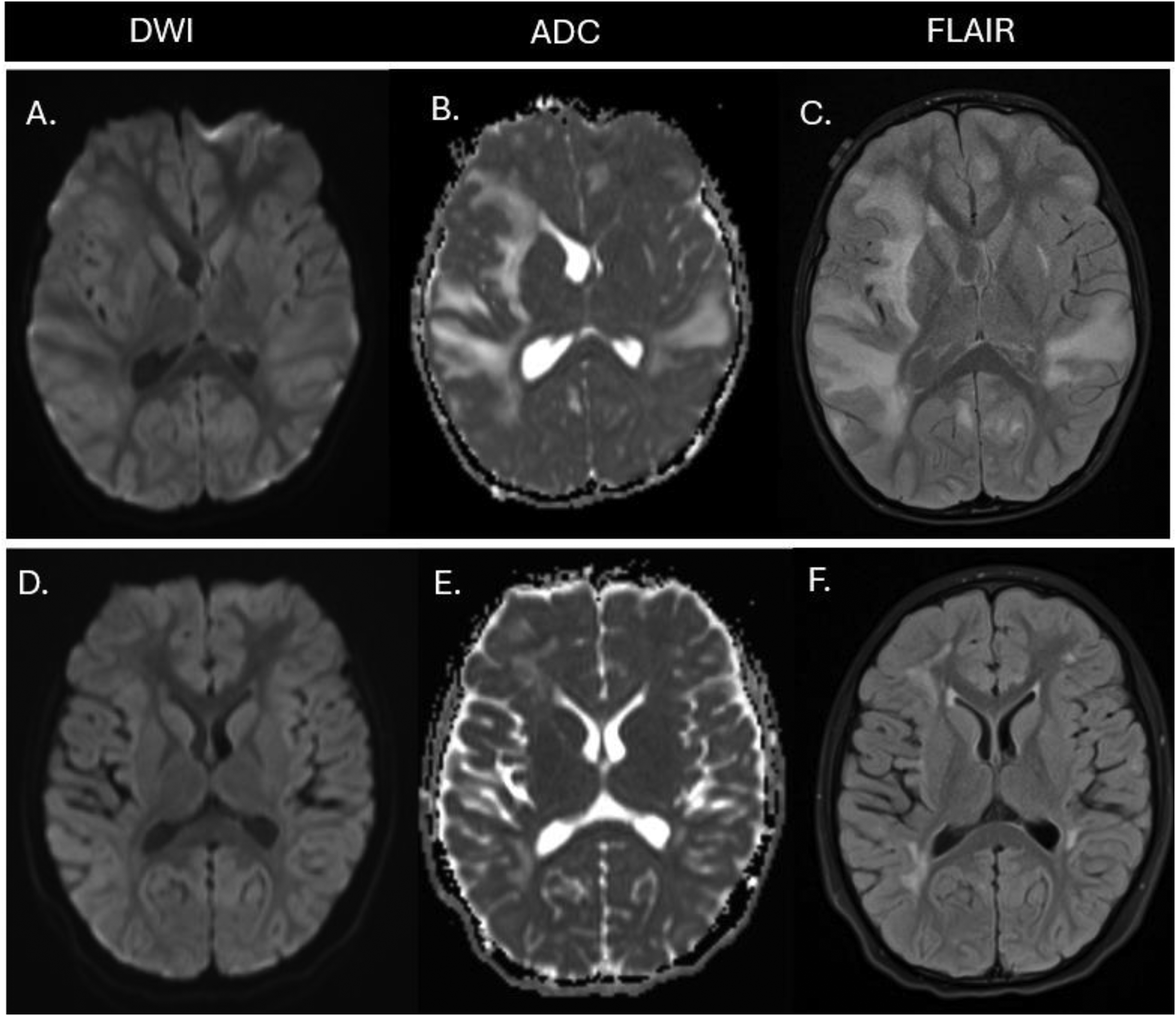
Supplemental Material
sj-docx-1-jcn-10.1177_08830738261456899 - Supplemental material for Acute Encephalopathy From Central Nervous System Hemophagocytic Lymphohistiocytosis (CNS HLH) in X-Linked Lymphoproliferative Disease Type 1 (XLP-1)
Supplemental material, sj-docx-1-jcn-10.1177_08830738261456899 for Acute Encephalopathy From Central Nervous System Hemophagocytic Lymphohistiocytosis (CNS HLH) in X-Linked Lymphoproliferative Disease Type 1 (XLP-1) by Quynh Kieu, Christine Le, Rishikesh Chavan and John Crawford in Journal of Child Neurology
Footnotes
Acknowledgments
We would like to acknowledge the support of CHOC hospital for providing the necessary resources and facilities for this study.
Ethical Approval
Our institution does not require ethical approval for reporting individual cases or case series.
Informed Consent
Written informed consent was obtained from the patient and their parents for the publication of their anonymized information in this article.
Author Contributions
Dr. Kieu and Dr. Crawford contributed to study conception and design, data analysis, and drafting of the manuscript. Dr. Le and Dr. Chavan contributed to data collection and provided critical review and editing of the manuscript. All authors approved the final version.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
This statement aligns with the guidelines provided in the sources, ensuring clarity and transparency
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.