
Abstract
Combined immunodeficiency (CID) due to ORAI1 deficiency is an ultrarare autosomal recessive disorder characterized by the coexistence of recurrent infections, ectodermal dysplasia, autoimmunity, and myopathy. Untreated patients die in early childhood due to recurrent severe infections. This report presents a familial case of CID due to ORAI1 deficiency, caused by a homozygous loss-of-function variant c.581T>C in the ORAI1 gene. The oldest of the siblings died because of recurrent severe infections in the third year of life. His brother survived because of hematopoietic cell transplantation (HCT) but developed other symptoms of the disease. The patient achieved the ability to walk unaided at age 18 months, with a waddling gait from the beginning. His neurologic status was stable in early childhood, with mild deterioration after age 6 years. At the age of 11 years, proximal muscle weakness and borderline respiratory sufficiency are observed, with myopathic electromyography, normal level of creatine kinase, and global fatty infiltration in muscle magnetic resonance imaging, most pronounced in gluteus maximus. Additionally, dental dysplasia and heat intolerance are observed. Keywords: ORAI1 deficiency, combined immunodeficiency, ORAI1 gene, neuromuscular symptoms, myopathy
Case report
An 11-year-old boy was the third child of consanguineous healthy parents (second cousins) (Figure 1). His oldest brother (II:1) had a severe combined immunodeficiency of unknown genetic origin. He was born after an uneventful pregnancy and had an Apgar 10. No clinical symptoms were observed in the first 6 months, except mild upper respiratory tract infection in the neonatal period. He was thriving, gaining weight. First pneumonia, complicated by diarrhea, was observed in the sixth month of age. At 9 months, he developed his second pneumonia, complicated by sepsis. Since then, he suffered from continuous upper and lower respiratory tract infections of different etiology, that is, pneumocystis as well as BCGitis. Despite recurrent infections, the boy achieved milestones on time. However, hypotonia and waddling gait were observed early, but not investigated due to the patient's general bad condition. He died at the age of 3 years, the ninth day after the hematopoietic cell transplantation (HCT), due to severe graft-vs-host disease (GvHD). The second brother (II:2) is unaffected. After his birth, the mesenchymal umbilical cord blood was banked. The presented patient (II:3) was born at 37 weeks of gestation, with an Apgar score of 9, weighing 2850 g (50th-90th percentile). The neonatal period was uneventful. The screening for immunologic deficits (lymphocyte subpopulations) did not reveal any deviations at that time. At the age of four months, the boy developed fever, bronchitis, and mild diarrhea with blood in stools. Initial blood analysis revealed anemia with leukocytosis and high IgE levels (>1000 IU/mL). Further immunologic examination showed hypergammaglobulinemia, IgE >2500 IU/mL, and progressive disturbances in T lymphocyte subsets. He was diagnosed with severe combined immunodeficiency. The patient underwent marrow transplantation at the age of 7 months, using umbilical cord blood from his healthy brother. After the transplant, he developed acute GvHD of the skin, grade II/III, which was successfully treated with steroids. Immunosuppressive treatment was discontinued after 2 months, with no symptoms of GvHD since then. However, because of increasing chimerism, the patient received donor lymphocyte infusions twice (at the age of 11 and 12 months). Despite the medical challenges, the boy achieved milestones with only mild delay: sitting at 9 months and walking at 18 months. Cognitive development was normal. However, from the beginning, he showed reduced motor skills, with waddling gait and difficulties climbing stairs. He's never been able to run. He was stable with ability to walk unaided and jump until around 6-7 years of age when his mother noticed a progression of motor symptoms and fatiguability. He had hypotonia, a waddling gait and Gowers sign. Additionally, sweating abnormalities were observed with reduced perspiration in high temperatures. Physical examination at the age of 9 revealed divergent strabismus, weak response to mydriatics, high-degree hyperopia and astigmatism, loss of enamel in teeth, normal hair (scalp, eyebrows, eyelashes) and skin, joint laxity (hyperextension in the elbows, knees, valgus feet), proximal weakness, more pronounced in the lower limbs, waddling gait and difficulty rising from a squatting position, necessitating the Gowers maneuver (Figure 1). The deep tendon reflexes were normal. During subsequent clinical follow-ups (at ages 10,11,13,14), there was a progression of symptoms, with slight deterioration confirmed with physiotherapy scales—with the results accordingly 371 m, 437 m, 291 m, and 360 m in 6-minute walk test and scores 16/34, 8/34 and 15/34 in Natural Sciences Admissions Assessment (NSAA) at ages 11, 13, and 14 years (test not performed at the age 10 years). Test results indicate progression of proximal limb muscle weakness (acc. MRC 4/5 in upper limbs: flexion, extension, abduction and adduction in the shoulder joints; and 3/5 in lower limbs: flexion, extension, abduction and adduction in the hip joints). Patient maintains ability to walk unaided, but he cannot run or jump. The follow-up at age 13 years was preceded by upper respiratory tract infection, which could have mildly affected the assessment.
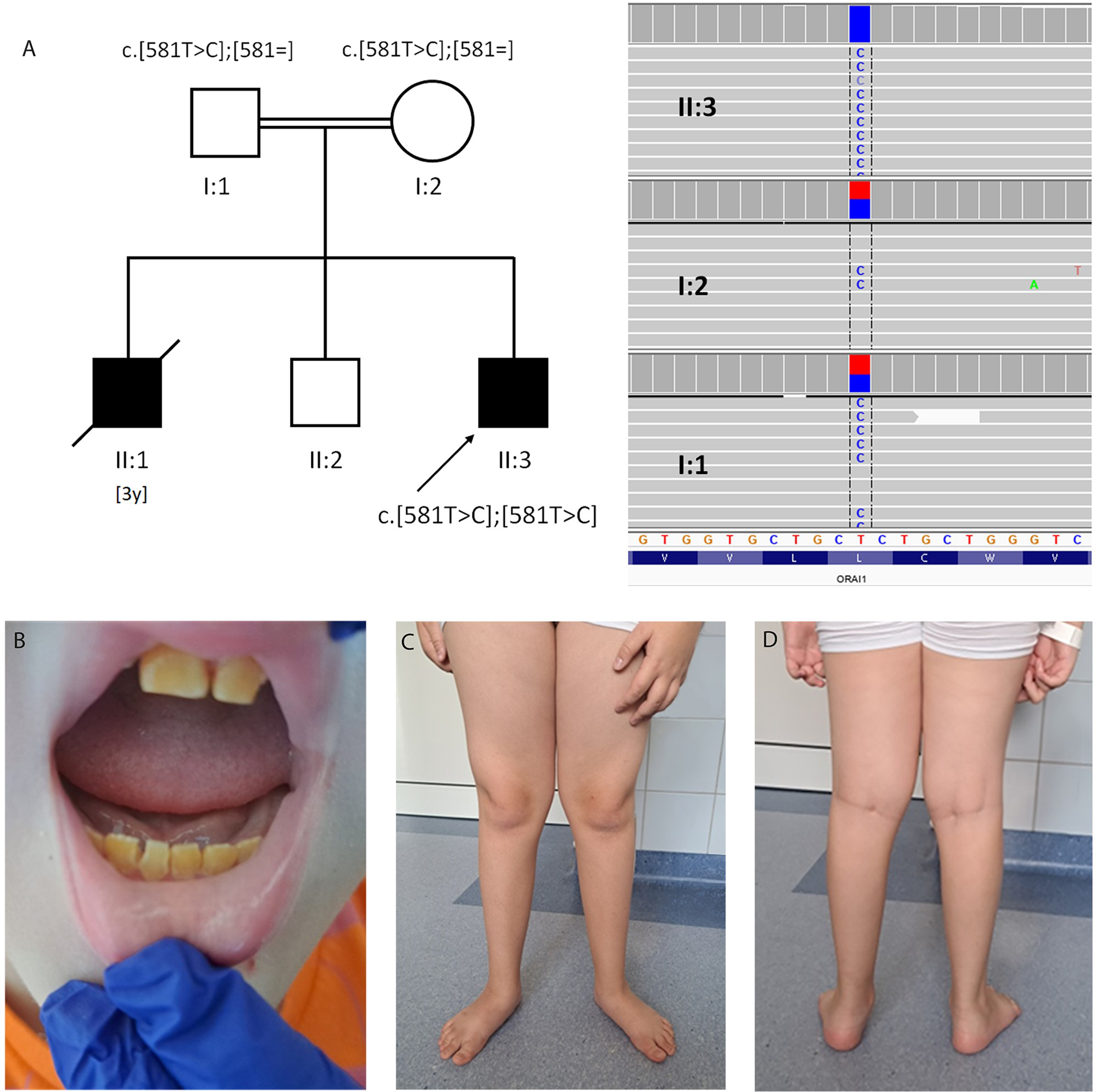
(A) Pedigree and Integrative Genomics Viewer (IGV) presentation of next-generation sequencing of the presented family. Parents are second cousins (I:1, I:2), and 2 sons (II:1, II:3) were affected. (B-D) Proband (II:3) at the age of 11 years. Note loss of enamel (B), hypotonia signs (pes and knee valgus) and distal atrophy (C, D).
Creatine kinase levels were normal.
Electrophysiological studies included nerve conduction studies, needle electromyography, and repetitive nerve stimulation. Nerve conduction studies demonstrated a slight reduction in compound muscle action potential amplitudes in the bilateral tibial nerves, whereas distal latencies and conduction velocities were within normal limits.
Needle electromyography of proximal muscles of the right upper and lower limbs (biceps brachii and vastus lateralis) revealed findings consistent with a primary myopathic pattern in biceps brachii, including marked abnormalities of motor unit recruitment with an early full interference pattern during voluntary contraction.
Repetitive nerve stimulation was performed at 3 Hz on the right radial nerve with recording from the anconeus muscle. The amplitude of the first compound muscle action potential was 2.9 mV. No decremental response (4:1 ratio) was observed, and no postexercise facilitation was detected.
Tables and figures from electrophysiological studies are enclosed in Supplementary Materials.
Magnetic resonance imaging (MRI) of the brain showed no abnormalities. Whole body MRI revealed diffuse symmetric fatty infiltration, severe in the gluteus maximus and moderate in the facial muscles, chest, abdominal wall muscles, upper limb muscles (more pronounced in the forearm than in the arm), paraspinal, and lower limb muscles. At the thigh level, the most visible changes were localized in the vastus muscles with relative sparing of the adductor magnus muscle. At the calf level moderate fatty infiltration presented in the soleus muscle, the lateral head of the gastrocnemius muscle, and peroneus muscles. No signs of edema were seen on short tau inversion recovery images (Figure 2). Pulmonary function tests showed restriction with forced vital capacity of 2.5 L. Polysomnography performed at the age of 13 years showed mild episodes of sleep apnea with no clinically significant carbon dioxide retention. No ventilation support has yet been applied. Densitometry revealed a transient generalized reduction in bone density at the age of 10 years, result of the test performed at the age of 13 years was within norm with a Z score of 1.0 (improvement achieved with vitamin D supplementation alone). Next-generation sequencing was employed with custom-designed panel. Genomic DNA was extracted from a dermal biopsy (due to HCT). Targeted sequencing of approximately 5 Mb of spanning exonic regions and exon-intron boundaries of more than 500 NMD-associated genes was conducted using KAPA HTP Library Preparation Kit (Roche NimbleGen) and paired-end (2 × 100 bp) sequenced on the NovaSeq6000 (Illumina) platform to the mean sample coverage 149× (ge10 = 95.9% and ge20 = 95.8%). The analysis revealed a homozygous variant NM_032790.4:c.581T>C (p.Leu194Pro) in the ORAI1 gene (12:121641318-T>C, GRCh38). Both parents were confirmed as carriers of the variant (Figure 1). We did not perform a muscle biopsy because of ethical reasons (invasive procedure).
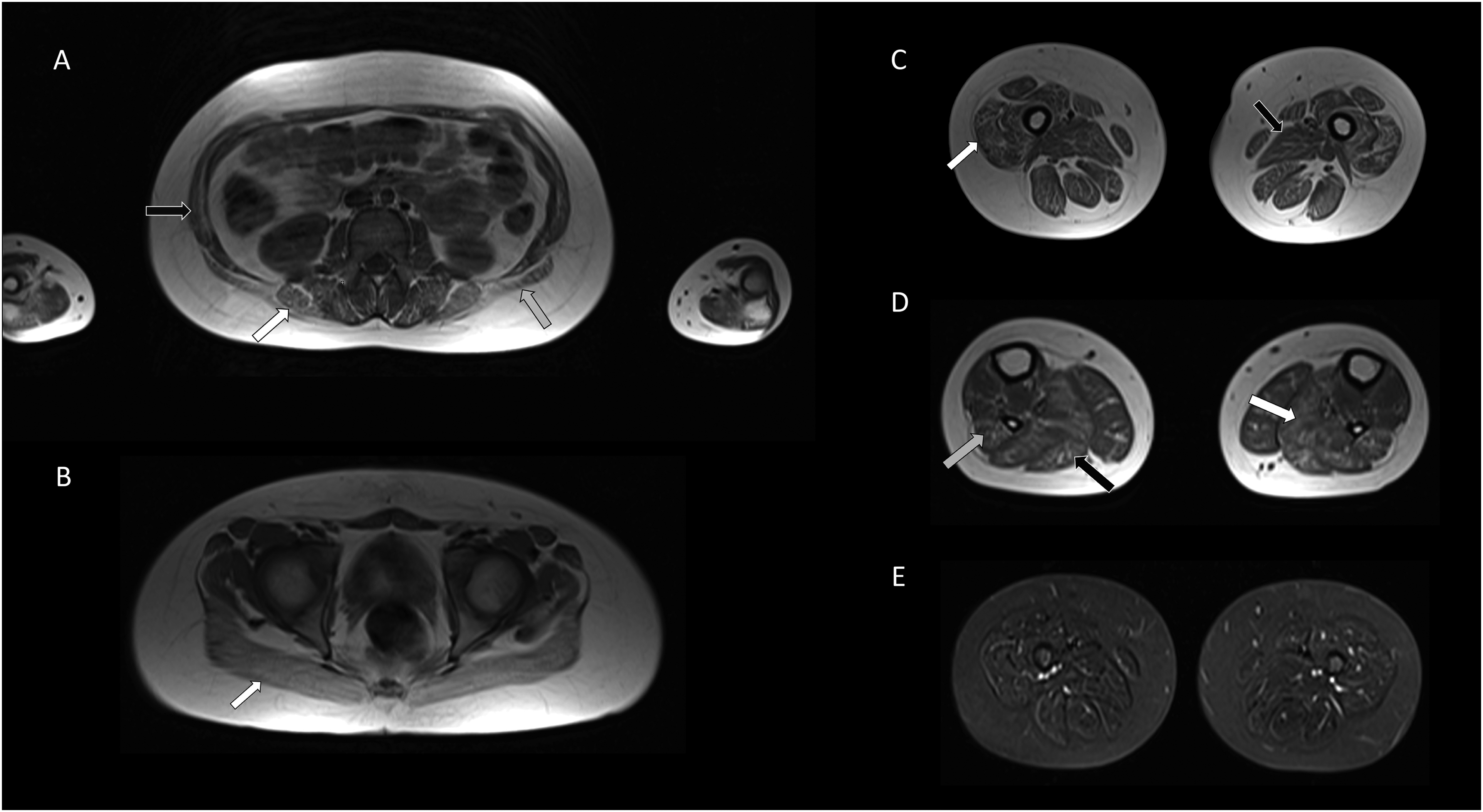
Whole body magnetic resonance imaging. (A) Moderate fatty infiltration in the lumbar paraspinal muscles (white arrow), abdominal wall muscles (black arrow), and latissimus dorsi muscle (gray arrow) (axial T1-weighted images). (B) Severely affected the gluteus maximus muscle (white arrow) (axial T1-weighted images). (C) At the thigh level diffuse fatty infiltration in all muscle compartments especially in the vastus muscles (white arrow) with relative sparing of the adductor magnus muscle (black arrow) (axial T1-weighted images). (D) At the calf level moderate changes in the soleus muscle (white arrow), the lateral head of the gastrocnemius muscle (black arrow), and peroneus muscles (gray arrow) (axial T1-weighted images). (E) No muscle edema in STIR images. STIR, short tau inversion recovery.
Discussion
Defects in T cell activation result in immunodeficiency associated with severe infections early in life. T-cell activation requires Ca2+ influx through calcium release–activated calcium (CRAC) channels. A plasma membrane protein encoded by the ORAI1 gene is essential for the CRAC channel function and is activated by Stromal Interaction Molecule 1 (STIM1), a key transmembrane protein that serves as a calcium level sensor.1–3 Loss-of-function variants ORAI1 and STIM1 genes impair CRAC channel activity, leading to combined immunodeficiency, including immunodeficiency 9 (ORAI1 deficiency; ORPHA: 317428, OMIM 612782) and immunodeficiency 10 (STIM1 deficiency; ORPHA: 317430 OMIM 612783), which are inherited in an autosomal recessive trait. In contrast, gain-of-function variants result in constitutive calcium influx and are typically inherited in an autosomal dominant manner. Gain-of-function variants in ORAI1 lead to tubular aggregate myopathy 2 (ORPHA: 2593, OMIM 615883), whereas gain-of-function variants in STIM1 cause Stormorken syndrome (ORPHA: 3204, OMIM 185070) and tubular aggregate myopathy 1 (ORPHA: 2593, OMIM 160565). Loss-of-function mutations in both genes affect a broad spectrum of cell types, including T, B, and NK lymphocytes, polymorphonuclear leukocytes, platelets, fibroblasts, skeletal muscle cells, eccrine sweat glands, and ameloblasts. In contrast, gain-of-function mutations predominantly affect skeletal muscle, whereas STIM1 gain-of-function variants additionally impact platelets. Compared with patients with STIM1 mutations—who frequently develop autoimmune diseases and lymphoproliferative disorders—patients with ORAI1 mutations primarily present with immunodeficiency, and autoimmune manifestations are rare (e.g. autoimmune hemolytic anemia, thrombocytopenia, splenomegaly, lymphadenopathy).2,4,5
Combined immunodeficiency due to ORAI1 deficiency is an ultra-rare disorder, characterized by life-threatening recurrent infections, accompanied by nonimmunologic symptoms, such as myopathy and ectodermal dysplasia,6,7 with only 18 patients reported to date.2,7–10 In all of them, severe immunodeficiency was observed in infancy, with recurrent life-threatening infections (such as chronic diarrhea, pneumonia, gastrointestinal sepsis, and BCGitis pyelonephritis) leading to death in infancy or early childhood. Among all reported cases, 7 patients survived following HCT. Available data suggest that HCT leads to long-term resolution of immunodeficiency-related symptoms in these patients. However, all individuals presented with hypotonia and a myopathic phenotype. McCarl et al 4 mentioned 2 patients alive at the age of 16 years after HCT with persisting myopathy, one of whom died at the age of 20 years as a result of respiratory failure. 2 Muscle biopsy in one of them showed atrophy of type II muscle fibers. Lian et al 7 presented a description of a 3.5-year-old patient, who showed considerable weakness in facial muscles and a Gowers sign. Electromyographic and nerve conduction studies performed at that time were normal. A muscle biopsy (gastrocnemius muscle) showed an almost complete absence of type II muscle fibers. At 6 years of age, this patient began to use a wheelchair intermittently. Our patient achieved motor milestones nearly on time; however, hypotonia and waddling gait were observed from the very beginning. Initially, these symptoms were attributed to the patient's general condition and complications after transplant. The concerns escalated after he turned 6 years old, as he started experiencing difficulties in getting up from a squatting position and climbing stairs. Since then mild deterioration in neurologic status has been observed, with global weakness more pronounced in lower limbs and proximal muscles. Additionally, 5 of 7 patients exhibited features of ectodermal dysplasia, also noticed in our patient. Other manifestations, such as corneal abnormalities, eczema, and hepatosplenomegaly, were reported in individual cases. Brain abnormalities on MRI were described in only 1 patient. All data are summarized in Table 1.
Patients With ORAI1-Related Immunodeficiency Who Underwent HCT.
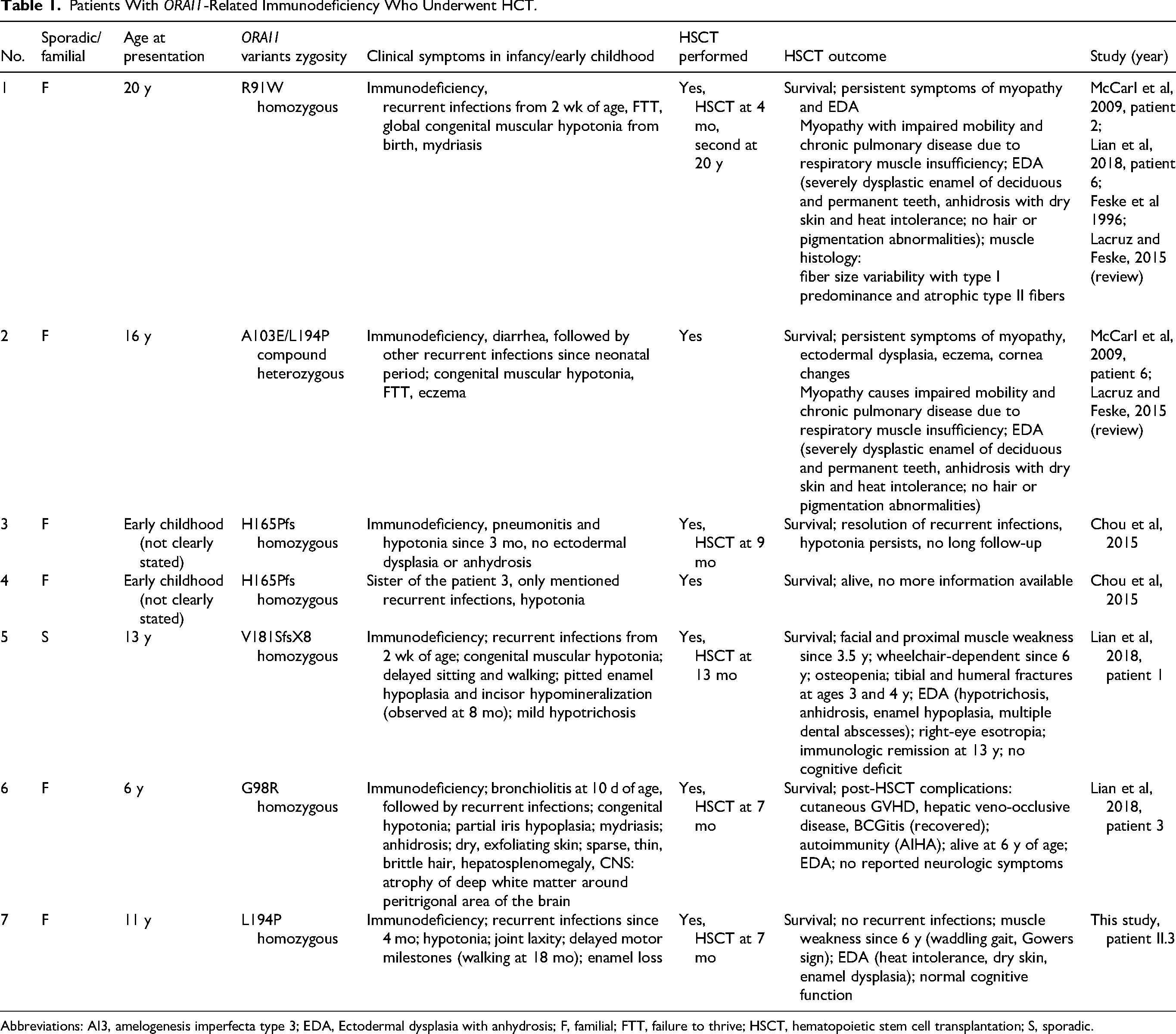
Abbreviations: AI3, amelogenesis imperfecta type 3; EDA, Ectodermal dysplasia with anhydrosis; F, familial; FTT, failure to thrive; HSCT, hematopoietic stem cell transplantation; S, sporadic.
In the index case, a homozygous single nucleotide transition c.581T>C in exon 2 of the ORAI1 gene was identified. This missense variant was previously described in two patients,4,7 once in compound hetero- and once in homozygous status.4,7 The patient with a homozygous variant (identical to our family) presented with a much more severe phenotype, compared with our patient. She suffered from severe infections since 2 months of age, congenital muscular hypotonia, poor head control, lacked spontaneous movements and facial mimicking, and respiratory failure. She had mydriasis with very slow pupil reactions to light. A biopsy from the deltoid muscle at 6 months of age showed variable fiber size but was otherwise normally structured. Despite intensive care, the girl died at 7.5 months of age. The c.581T>C variant results in a missense substitution (p.Leu194Pro), involving the alteration of a nonconserved nucleotide. According to the automated ACMG/AMP classification implemented in the GeneBe platform (PMID: 38440907), this variant has been classified as pathogenic (14 points; PS3, strong; PP3, moderate; PP5, very strong). The c.581T>C variant in the ORAI1 gene has an allele frequency of 0.00001549 in the gnomAD v4.1.0 database (accessed March 18, 2026), with 25 heterozygous carriers and no reported homozygotes. Further evaluation of allele frequency across gnomAD subpopulations revealed that all heterozygous carriers are of European ancestry, predominantly non-Finnish European (23/25), which may suggest a potential founder effect within European populations. Functional studies showed that c.581T>C variant results in protein loss of function by severely compromising stable ORAI1 protein expression, abolishing Ca2+ influx, despite no effect on ORAI1 mRNA transcription.4,7
In summary, our patient presents with myopathy, with symptoms observed from early childhood and slow progression from the age of 6. Joint laxity and nonprogressive symptoms of ectodermal dysplasia are the predominant accompanying features. We did not observe autoimmunity. Although HCT saved the child's life, it did not protect against the neurologic symptoms of the disease. The prognosis is currently unclear with risk of chronic respiratory insufficiency. There is a need for reporting similar cases to better understand the natural history of the disease.
Supplemental Material
sj-pdf-1-jcn-10.1177_08830738261459493 - Supplemental material for Neuromuscular Symptoms of ORAI1-Related Immunodeficiency
Supplemental material, sj-pdf-1-jcn-10.1177_08830738261459493 for Neuromuscular Symptoms of ORAI1-Related Immunodeficiency by Maria Jędrzejowska, Anna Potulska-Chromik, Karolina Czeczko, Maria Franaszczyk, Beata Wolska-Kuśnierz, Edyta Rosiak, Karolina Aragon-Gawińska, Tomasz Stokłosa and Anna Kostera-Pruszczyk in Journal of Child Neurology
Footnotes
Acknowledgments
We thank the patient and his family members for their participation in this study. In addition, we thank the team of physiotherapists from our clinic, headed by Malgorzata Burlewicz.
ORCID iDs
Informed Consent
Parents of the child provided written informed consent for clinical, laboratory, and genetic testing. The study was approved by the local ethical committee of the Medical University of Warsaw.
Consent for Publication
Written informed consent for publication of the patient's clinical data and images was obtained from the parents.
Author Contributions
M Jędrzejowska, A Potulska-Chromik, K Czeczko, K Aragon-Gawińska, B Wolska-Kuśnierz, A Kostera-Pruszczyk – development of medical history materials, literature review; E Rosiak –magnetic resonance imaging study with critical literature review; M Jędrzejowska, M Franaszczyk, T Stokłosa – genetic testing, critical literature review; M Jędrzejowska, K Czeczko, A Potulska-Chromik, A Kostera-Pruszczyk – study design, preparation of the manuscript, literature review. All co-authors: critical review of the final manuscript
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Sanofi Genzyme (grant number Genzyme-Sanofi no 1WC/DAR3/16 UKI/343, UH000123-05/16).
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.