
Abstract
The development of self-assembled nanocarriers for the encapsulation of hydrophobic antioxidants is of growing interest. Self-assembled amphiphilic chitosan conjugate nanocarriers that stabilize antioxidants were prepared based on the concept that both the nanocarrier and the antioxidant bear similar hydrophobic moieties able to establish hydrophobic interactions. This work describes the preparation and characterization of a system consisting of a palmitoyl chitosan conjugate and retinyl palmitate. Palmitic acid was coupled to chitosan using a carbodiimide-mediated coupling reaction, and two different palmitoyl chitosan conjugates were obtained by varying the coupling system. Palmitoyl chitosan conjugates self-assembled to form nanoparticles in aqueous medium varying in mean average diameter (Dh) between 200 and 437 nm. Retinyl palmitate–loaded nanoparticles were prepared by a solvent displacement method using dialysis, with loading efficiencies of 77.5% and 88.6%, loading contents of 12.6% and 14.6%, and Dh values of approximately 280 nm. The zeta potential (ζ) of all palmitoyl chitosan nanoparticle were above 25 mV, but ζ slightly increased in the retinyl palmitate–loaded nanoparticle. Antioxidant activity of loaded nanoparticles was confirmed using the 1,1-diphenyl-2-picryl-hydrazyl radical scavenging assay. The in vitro cytotoxicity of blank and loaded nanoparticles was determined using fibroblasts of human embryonic skin. All nanoparticles were not cytotoxic when they were tested with methylthiazol tetrazolium and lactate dehydrogenase tests. The obtained results suggest that the system has potential as a nanocarrier for dermal application. Additionally, the approach considered in this article can be expanded to other nanocarrier/antioxidant systems.
Introduction
Vitamin A (retinol) and its derivatives1,2 have a recognized potential in skin care, 3 that is, antiaging and prevention of inflammation and skin diseases. 4 The biological activities of vitamin A include thickening of the epidermis, 5 increasing the autolysis of keratinocytes, 6 glycogen deposition, and synthesis of collagen and elastin.7,8 One important limitation of vitamin A in dermal tissue is the occurrence of local irritation such as erythema, xerosis, and mild scaling. 9 Another drawback of vitamin A is its instability when exposed to light or heat. 10 The use of derivatives such as esters11,12 has somewhat counteracted these limitations. Research continues to focus on the development of nanoaggregate systems able to encapsulate or conjugate the retinoid and improve its poor solubility along with its biocompatibility. 13 Particularly, research on self-assembled nanoparticles (NPs) is increasing. Macromolecular self-assembly is a spontaneous process that leads to the formation of core-shell nanostructures with different morphologies such as micelles and NPs. This process is frequently achieved by amphiphilic polymers with the inner core consisting of the hydrophobic part of the polymer and serving as a nanocontainer for poorly soluble molecules. This core is surrounded by an outer shell of hydrophilic blocks or segments of the amphiphile.14,15
Recently, attention centered on the preparation of nanocarriers based on the self-assembly of amphiphilic polysaccharides. 16 Among them, chitosan (Ch), the N-deacetylated derivative of chitin (poly-β-(1→4)-N-acetyl-D-glucosamine), has been extensively used due to its unique and versatile physico-chemical and biological properties.17–20 Many amphiphilic derivatives of chitosan have been reported in the literature and are based on the conjugation with alkyl residues,21,22 fatty acid moieties,23,24 nonlinear hydrophobic molecules25,26 and even hydrophobic or hydrotropic molecules.27,28 In particular, for the delivery of antioxidants, several colloidal, amphiphilically modified chitosans have been proposed as nanocarriers, and chitosan can also contribute to the radical scavenging activity (RSA) due to its own recognized antioxidant properties.29,30 Several systems contained chitosan with retinol or all-trans retinoic acid (ATRA) since both retinoids are able to form H– bonds with the polysaccharide.31,32 Based on this concept, retinol has been encapsulated in water-soluble succinated chitosan NP 33 and ATRA has been physically entrapped inside self-assembled NP of 6-O-cholesterol-modified chitosan conjugates. 34 In other works, ATRA was chemically linked to oligomeric chitosan, and this amphiphile self-assembled successfully into micelles. 27 Following another pathway, ATRA was incorporated into NPs of methoxy poly(ethylene glycol)-grafted chitosan through ion-complex formation between ATRA and chitosan. 35 This work proposes the development of an antioxidant nanosystem using a new approach based on the fact that both nanocarrier and antioxidant bear similar hydrophobic moieties able to establish hydrophobic interactions. Due to the affinity and simularity of the chemical structures of the hydrophobic residues present in both the amphiphile and the antioxidant, an enhanced loading efficiency (LE) is expected.
This article describes the preparation and characterization of a system consisting of a palmitoyl chitosan (Ch/P) conjugate containing entrapped retinyl palmitate (RP). RP was selected because of its enhanced biocompatibility as compared to retinol and ATRA. 36 Palmitoyl chitosan conjugates were prepared by using two different synthetic conditions, and their physico-chemical properties were analyzed. Self-assembled NPs of these amphiphiles encapsulating RP were obtained in aqueous medium. NPs were characterized by Fourier transform infrared spectra (FTIR) and elemental analysis. Particle size distributions and zeta potential of NPs were determined by dynamic light scattering (DLS), and the morphology was examined by microscopic techniques. In addition, the antioxidant activity of RP-loaded NPs was assessed by 1,1-diphenyl-2-picryl-hydrazyl (DPPH•) radical scavenging assay, and their biocompatibility was evaluated with fibroblasts of human embryonic skin (HFB) using standard assays.
Materials and methods
Materials
Water-soluble chitosan chloride (Ch, same abbreviation as chitosan; Protasan UP CL 213, NovaMatrix), palmitic acid (PA, Sigma–Aldrich), 1-(3-dimethyl-aminopropyl)-3-ethylcarbodiimide (EDC; Agilent Technologies), N-hydroxysuccinimide (NHS; Sigma–Aldrich), 37% hydrochloric acid (HCl; VWR Chemicals), sodium hydroxide (NaOH; Panreac), potassium phthalate (KHP; Riedel-de-Haën), potassium bromide FTIR grade (KBr; Sigma–Aldrich), RP (Sigma–Aldrich), DPPH• (Sigma–Aldrich), ethanol (VWR Chemicals), dimethylsulfoxide (DMSO; Scharlab) and phosphate-buffered saline (PBS) solution, pH 7.4 (Sigma–Aldrich) were used as received.
Preparation of Ch/P conjugates
Procedure 1
Based on an NH2:COOH mass ratio of 10:1, chitosan (500 mg) was dissolved in deionized water (200 mL) and PA (50 mg) was dissolved in ethanol (100 mL). The EDC (350 mg, EDC:PA molar ratio of 7:1) was dissolved in deionized water (25 mL). All three solutions were mixed at 80°C, and the reaction was allowed to proceed for 18 h at 80°C under stirring. The reaction solution was dialyzed (molecular weight cut-off (MWCO) = 3500, Spectrum Laboratories Inc.) against a water:ethanol solution 50:50 v/v for 24 h and after that against deionized water for 48 h. The reaction product was isolated by freeze-drying (Telstar LyoQuest HT40 Beijer Electronics) for 24 h, giving a white powder which was labeled as Ch/P1.
Procedure 2
Based on an NH2:COOH mass ratio of 10:1, chitosan (100 mg) was dissolved in deionized water (10 mL) and PA (10 mg) was dissolved in DMSO (4 mL). The EDC (10.2 mg, EDC:PA molar ratio of 1.2:1) was dissolved in deionized water (2 mL) and NHS (4 mg, PA:NHS molar ratio of 1:0.8) was also dissolved in deionized water (2 mL) but separately. The four solutions were mixed under agitation at 45°C, and the reaction was allowed to proceed for 18 h at the same temperature. The reaction solution was dialyzed (MWCO = 3500, Spectrum Laboratories Inc.) for 72 h against deionized water. The reaction product was isolated by freeze-drying (Telstar LyoQuest HT40 Beijer Electronics) for 24 h, giving a white powder which was labeled as Ch/P2.
Preparation of Ch/P NP
Ch/P NPs were prepared by suspending the corresponding amount of the chitosan derivative in distilled water to achieve concentrations of 0.05, 0.1, and 0.5 mg/mL, and stirring the dispersion overnight. Then, the dispersion was sonicated (Sonics Vibra-Cell) for 20 min with 36% amplitude dial setting and finally filtered using an Acrodisc® 25-mm syringe filter with a 1–µm glass fiber membrane (PN4523T 50 PK Sigma–Aldrich). Dry NP was obtained by freeze-drying (Telstar LyoQuest HT40 Beijer Electronics) the aqueous dispersion for 24 h. These NPs were labeled Ch/P1 NP and Ch/P2 NP according to the chitosan conjugate used.
RP-loaded Ch/P NP were prepared by solvent displacement using dialysis under lightproof conditions. RP (10 mg) was dissolved in DMSO (10 mL), and this solution was dropped into an aqueous dispersion of Ch/P (50 mg) in deionized water (100 mL) under agitation using a homogenizer (IKA T 25D ULTRA-TURRAX, Laboratory EQUIPMENT, Germany) at 7200 rpm, at room temperature. After adding the RP solution, agitation was maintained for 30 min, and then sonication (36% amplitude dial setting) was applied for 20 min. The final dispersion was dialyzed (MWCO = 3500, Spectrum Laboratories Inc.) against water for 72 h. Dry NPs were obtained by freeze-drying (Telstar LyoQuest HT40 Beijer Electronics) for 24 h. These NPs were labeled RP-loaded Ch/P1 NP and RP-loaded Ch/P2 NP according to the palmitoyl Ch conjugate. Aqueous dispersions of RP-loaded NPs at 0.05, 0.1, and 0.5 mg/mL were obtained by suspending the NPs in distilled water applying the same procedure described above for obtaining Ch/P NP from their powders.
Physico-chemical characterization
Chemical analysis
The FTIR spectra were recorded on a Perkin–Elmer Spectrum One spectrophotometer. Potassium bromide was used to prepare slabs. Nitrogen content (N%) and carbon content (C%) were measured with a LECO CHNS-932 elemental analyzer using an infrared (IR) detector. Elemental analysis was used to quantitatively analyze starting materials, Ch/P derivatives, and RP-loaded Ch/P NP samples. This method was applied to determine the degree of substitution (DS, defined as the amount of palmitoyl moieties per 100 glucosamine units of chitosan) in the Ch/P derivative samples37–39 and to determine the drug loading efficiency (LE; defined as the percentage of RP in the NPs and the total amount of RP) and drug loading content (LC; defined as the percentage of RP in the NPs and the total amount of NP weight) 40 in the NP samples.
Determination of free amino groups
The amount of amino groups in the amphiphilic Ch derivatives was measured using a pH titration method previously reported. 41 Briefly, 0.2 g of sample was added to 25 mL of 0.1 N HCl solution, which was titrated with 0.1 N NaOH previously normalized with KHP.
Particle size distribution and zeta potential
The particle size distribution and the zeta potential of the NPs were determined by DLS using a Malvern Nanosizer Nano ZS Instrument equipped with a 4 mW He–Ne laser (λ = 633 nm) at an angle of 173°. Measurements of NP suspensions varying in concentration between 0.05 and 0.50 mg/mL were performed in square polystyrene cuvettes (SARSTEDT). The particle size distributions and the apparent hydrodynamic diameter (Dh) were obtained in terms of intensity. The zeta potential was determined for NP suspensions at 0.1 mg/mL concentration containing 2 mM NaCl 42 and using laser Doppler electrophoresis (LDE) with 30 runs/measurement. The temperature was kept constant at 25°C during size and zeta potential measurements. The zeta potentials were automatically calculated from the electrophoretic mobility using the Smoluchowski approximation.
Morphological analysis
The scanning electron microscopy (SEM) examination was performed using a Hitachi SU8000 FE-SEM apparatus at an accelerating voltage of 2.0 kV.
Transmission electron microscopy (TEM) images were recorded with a Hitachi SU8000 microscope operating at an accelerating voltage of 30 kV. A drop of the corresponding NP suspension was applied to carbon-coated copper grids, blotted, washed, and negatively stained with 2% (w/v) of phosphotungstic acid, and then air dried. Samples were diluted 20- to 200-fold when necessary before deposition on the grids.
Atomic force microscopy (AFM) was performed in tapping mode using a Multimode AFM (Veeco Instruments) equipped with a Nanoscope IVa control system (software version 6.14r1). Silicon-tapping probes (RTESP, Veeco) were used with a resonance frequency ~300 kHz and a scan rate of 0.5 Hz. For each sample, 10 × 10 µm2, 5 × 5 µm2, and 2 × 2 µm2 topographical AFM images were taken.
Thermal characterization
The thermogravimetric data were obtained using a thermogravimetric analyzer (TGA) using a TGA Q500 (TA instruments) apparatus, under dynamic nitrogen at a heating rate of 10°C/min in a range of 30°C–600°C.
Biological activity bioassays
In vitro antioxidant ability
The radical scavenging activity (RSA) of RP was evaluated by DPPH• experiments. In this experiment, a stock solution of RP in ethanol (41.7 mM) was prepared and sequentially diluted. Then, 1 mL of each RP dilution was mixed with 1 mL of DPPH• solution in ethanol (0.127 mM), and the decrease in absorbance at 515 nm was monitored every 30 min using a BioTek Synergy HT detector. The RSA was calculated using equation (1)
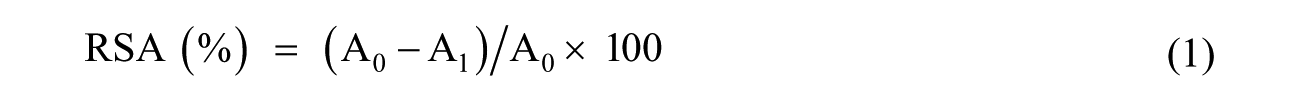
where A0 and A1 correspond to the absorbance at 515 nm of the radical DPPH• in the absence and presence of antioxidant, respectively.
Blank (unloaded) NPs and RP-loaded NPs of both conjugates were dispersed in ethanol (10 mg/mL) and stored in the dark for 2 days. Then, 1.0 mL of DPPH• ethanolic solution (0.127 mM) was mixed with 1.0 mL of the ethanolic extracts of NPs and the RSA monitored over time was determined by equation (1).
Cell culture and cytotoxicity
The biological response to the materials was tested with fibroblasts of human embryonic skin (HFB, Innoprot). The culture medium was Dulbecco’s modified Eagle’s medium modified with HEPES and enriched with 4500 mg/ml glucose (DMEM, Sigma) supplemented with 10% fetal bovine serum (FBS), 200 mM
For the evaluation of the cytotoxicity of NPs by methylthiazol tetrazolium (MTT) assays, cells were seeded at a density of 1 × 105 cells/mL of complete medium and were incubated to confluence in 96-well plates. After 24 h of incubation, the medium was replaced with the corresponding NP dispersion and incubated at 37°C in humidified air with 5% CO2 for 3, 6, and 13 h. 100 microliters of MTT solution was added to each well and the plates were incubated at 37ºC for 4 h. Excess medium and MTT were removed, and 100 µL DMSO was added to all wells. After mixing for 10 min, the absorbance was measured with a BioTek Synergy HT Plate Reader using a test wavelength of 570 nm and a reference wavelength of 630 nm. The percentage of relative cell viability (CV) was calculated using equation (2)
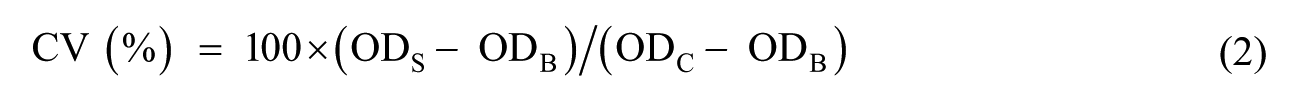
where ODS, ODB, and ODC are the optical densities of formazan for the sample, the blank, and the control, respectively.
For the evaluation of the cytotoxicity by lactate dehydrogenase (LDH) assays, cells were seeded at a density of 9 × 104 cell/mL. Then, NPs were added, and cells were incubated for 3, 6, and 13 h. The 96-well plates were shaken briefly to homogenize the LDH released, and the medium was recovered and conserved at −20°C. At the same time, the cells were lysed by a solution of triton X-100 in PBS (0.1%) for 30 min and then centrifuged at 5000 rpm and 4°C for 10 min. The supernatant was collected for further treatment. After collecting all the samples, 25 µL of the medium and supernatant were added into each well of the 96-well plate (n = 8). Next, 50 µL of the reaction mixture (TOX-7, Sigma–Aldrich) was also added into each well, and the plates were incubated for 30 min at room temperature in the dark, and 10 µL of 1 N HCl was added to each well to stop the reaction. Finally, the absorbance was measured at 490 nm, with a reference wavelength of 630 nm using a BioTek Synergy HT Plate Reader. The percentage of relative LDH release was determined with respect to control.
Cellular uptake
RP-loaded Ch/P2 NPs were prepared in PBS (50 µg/mL) in the presence of coumarin-6 (0.01%) (Sigma–Aldrich) by applying the same procedure described in section ‘Preparation of Ch/P NP’. Fibroblasts were seeded (2 × 104 cells/mL) in complete medium, and cells were incubated overnight at 37°C. Afterward, the medium was replaced with 500 µL of test sample and incubated for 6 and 18 h at 37°C. After each period, the cells were washed three times with cold PBS and fixed by a paraformaldehyde solution in PBS (3.7% w/v) for 10 min at 37°C, permeabilized with 0.5% triton in PBS for 30 min and finally washed twice with PBS. The nuclei were stained with the blue fluorescent dye Hoechst 33342 (1 µL/1000 µL in PBS, Sigma–Aldrich), and actin filaments were stained with the red staining agent phalloidin (1 µL/100 µL in PBS, Sigma–Aldrich) and incubated for 30 min at room temperature. Then, samples were washed with 0.1% Tween 20 in PBS and dried at room temperature. Cells were observed using epifluorescence microscopy with a Nikon Eclipse TE 2000-S fluorescence microscope system.
Statistical treatments and software
Statistical analysis was performed for MTT and LDH assays. ANNOVA analysis was performed at a significance level of 0.05 of the samples with respect to control using the SPSS software.
Results and discussion
Preparation and characterization of amphiphilic chitosan derivatives
The first objective of this work was the preparation of palmitoyl chitosan conjugates able to self-assemble into NPs as carriers of RP. The coupling reaction of chitosan was carried out using a water-soluble carbodiimide (EDC) following two routes: the first one involving the activation with EDC and high temperature (Ch/P1 derivative) and the second one, the activation with EDC/NHS at a mild temperature (Ch/P2 derivative). The preparation of amphiphiles of chitosan with palmitoyl residues is described in the literature employing N-acylation reactions with PA chloride 21 or with palmitic anhydride. 43 To our knowledge, the amidation reaction of PA and chitosan has not been described using EDC, however it was reported for the amidation of chitosan oligosaccharides with oleic acid,44,45 linoleic acid, 23 and stearic acid. 24 In this work, the coupling reaction of PA to chitosan mediated either by EDC or EDC/NHS proceeded successfully, reaching high reaction yields, with values of 89% and 95%, respectively. It was found that the yield was only slightly higher for the synthesis using EDC/NHS, which is considered to be more efficient in amidation reactions, 46 but the milder reaction conditions allowed for similar yields.
Shown in Figure 1 are the FTIR spectra of the Ch conjugates. The characteristic band of the palmitoyl residues at 2850 cm−1 was due to the stretching vibration of C–H in the CH3 and CH2 groups47,48 and the band at 1473 cm−1 was due to the deformation vibration of the same groups.
47
The band appearing at 2920 cm−1 was ascribed to the stretching vibration of C–H bonds in the chitosan as well as in the CH3 group of PA. The characteristic band due to the carboxylic group of PA at 1700 cm−1 disappeared, thus confirming the amidation reaction of chitosan. On the other hand, both spectra had the band that is attributed to the stretching vibration of the secondary amide C=O bond (amide I) (at 1629 and 1637 cm−1 for Ch/P1 and Ch/P2, respectively), and the band attributed to the N–H bending vibration in the
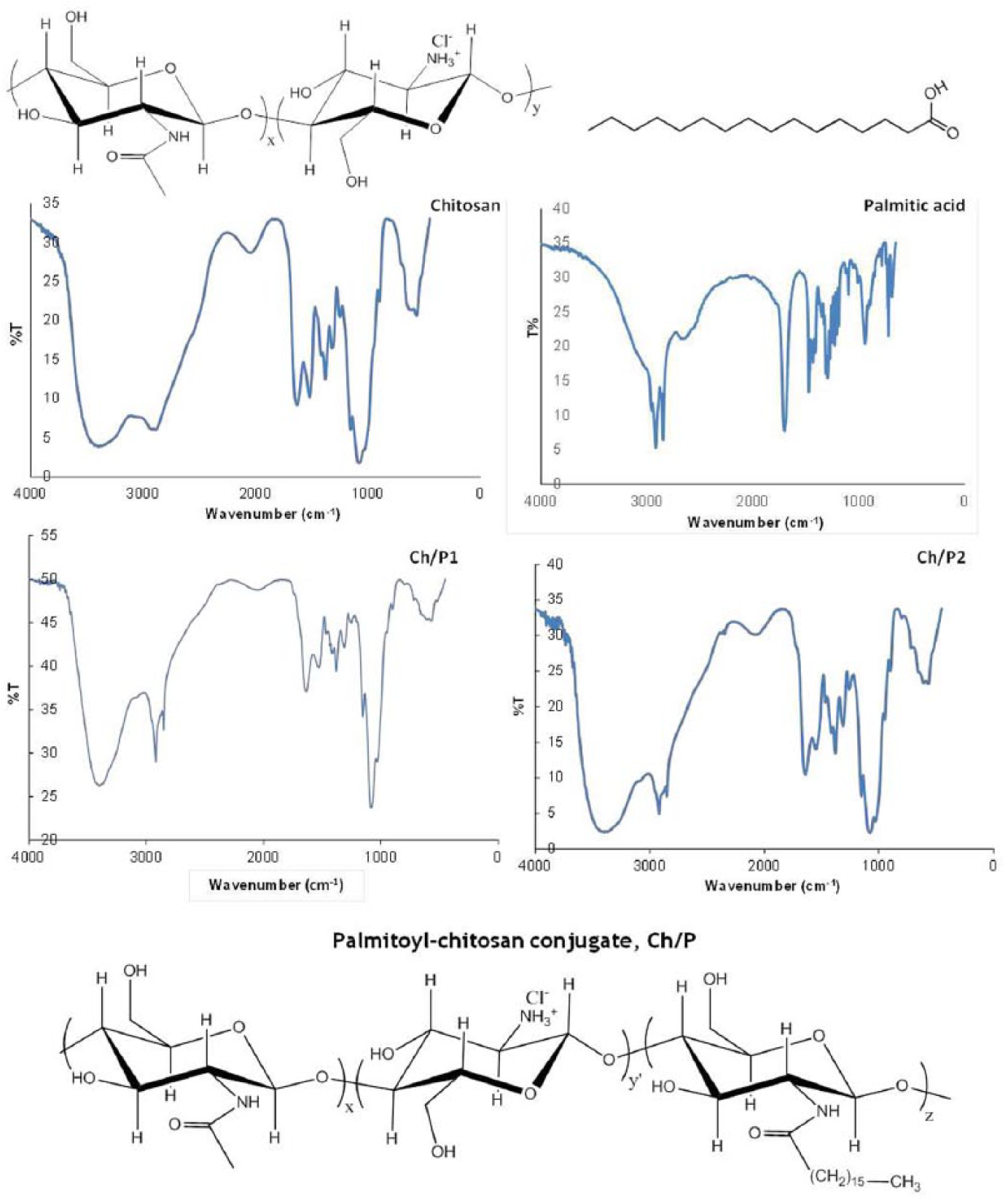
FTIR spectra of starting materials and Ch/P conjugates along with their chemical structures.
Results of elemental analysis of starting materials, Ch/P conjugates, and RP-loaded Ch/P NP.
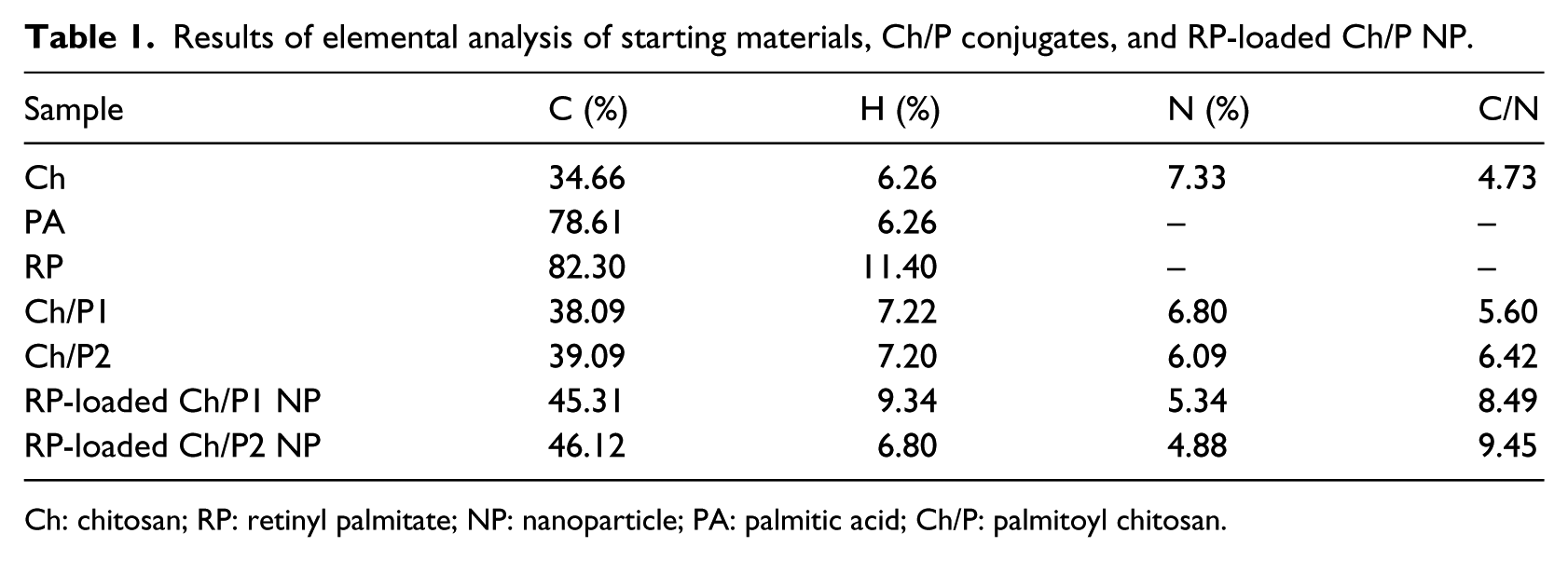
Ch: chitosan; RP: retinyl palmitate; NP: nanoparticle; PA: palmitic acid; Ch/P: palmitoyl chitosan.
Preparation and characterization of NPs
Blank (unloaded) and RP-loaded NPs of both Ch conjugates were obtained in aqueous medium. According to the polycore model described by Akiyoshi et al., 49 that proposes that the hydrophobic microdomains provides noncovalent crosslinks, and that have been applied to other amphiphilic chitosan systems, 50 our self-assembled NP would form by the association of the palmitoyl moieties into hydrophobic domains forming the NP core and the chitosan backbones coils forming the hydrophilic outside shells, to achieve a minimal energy state in aqueous medium. In addition, for loaded NPs, the hydrophobic interactions with the RP moieties would contribute to the formation of more compact cores.
RP-loaded NPs were analyzed by FTIR spectroscopy (Figure 2). The most significant finding was the appearance of a new and prominent band at 1738 cm−1 that was assigned to the stretching vibration of carbonyl in the ester group of RP, confirming the presence of the antioxidant in the NP. On the other hand, the bands of amide I and N–H bending vibrations in
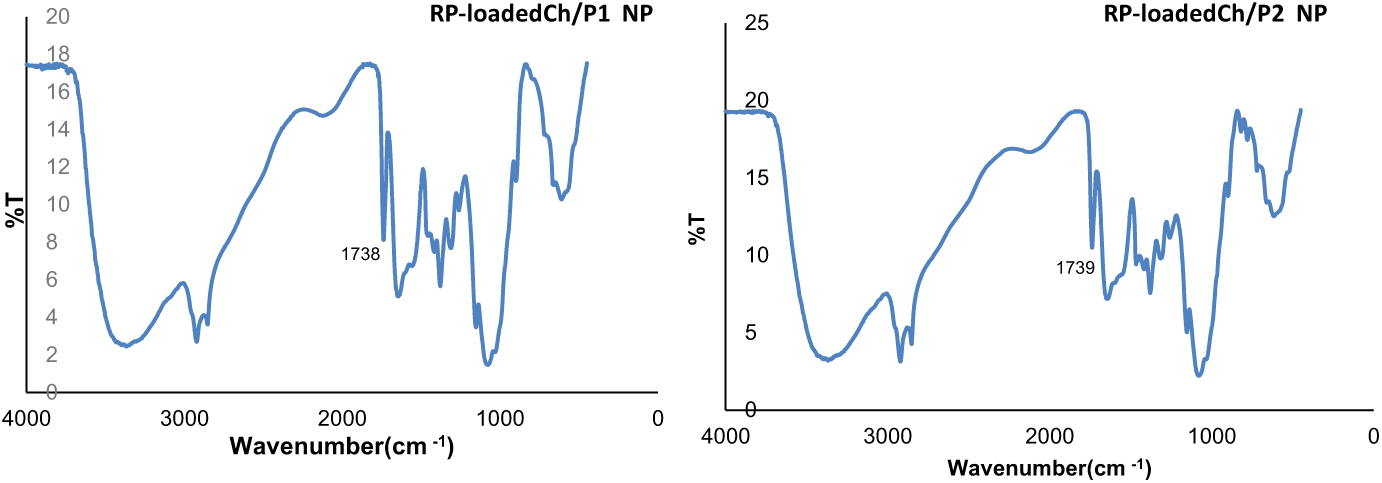
FTIR spectra of dried RP-loaded Ch/P NP.
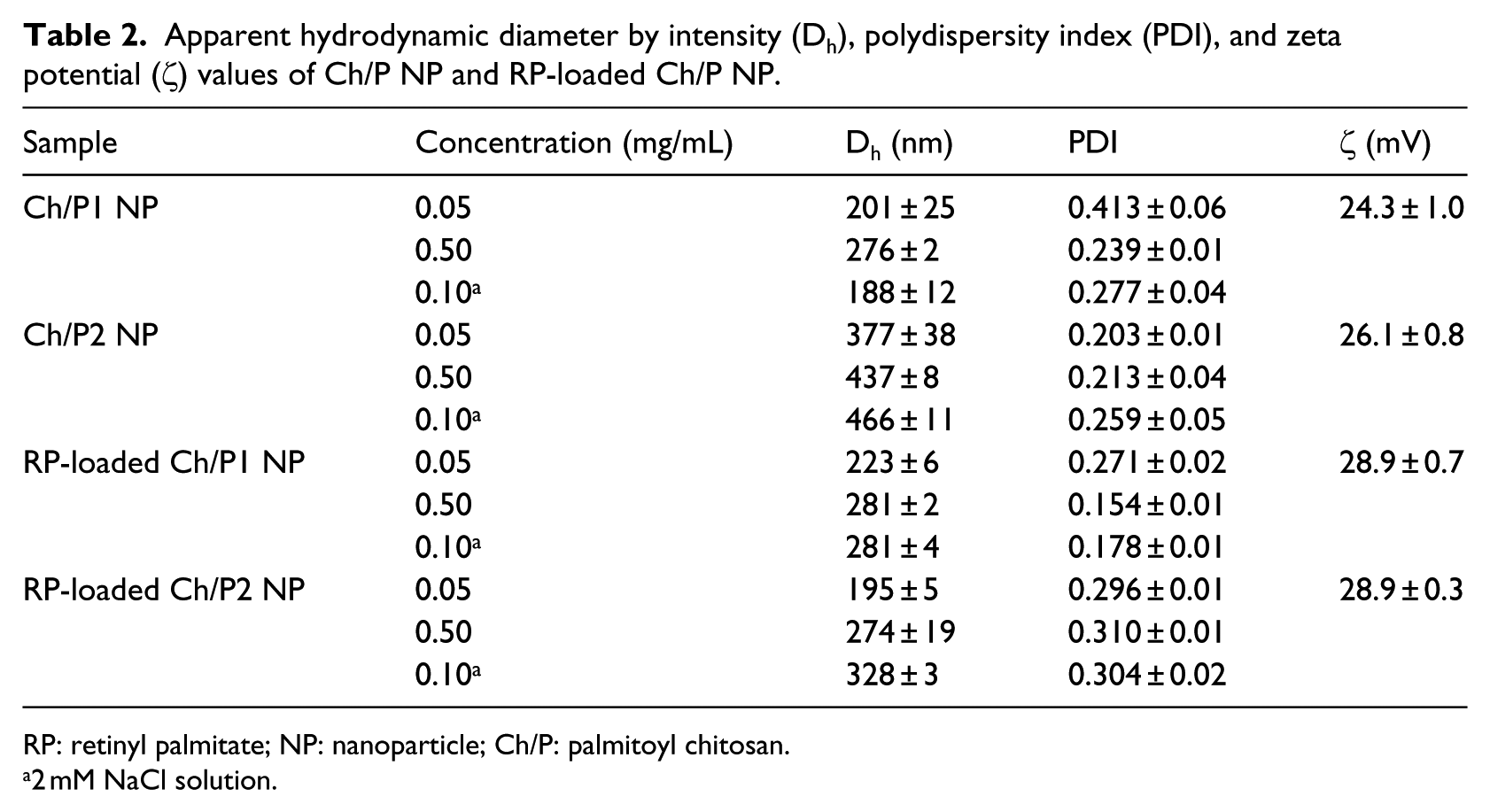
The particle size distributions were determined by DLS, and the Dh results are summarized in Table 2. Ch/P1 NP presented lower Dh values (almost half) than those of Ch/P2, at any concentration, suggesting a different self-assembly arrangement in each derivative. It is assumed that the modification of glucosamine cyclic structures of chitosan are most probably produced in a random way, 49 but depending on the distribution of the modified cycles along the polymer chains, the behavior of the modified polymers changes in the self-assembly process. In the case of the Ch/P2 system, the modified structures favor the formation of self-assembled agglomerated NPs giving an average diameter of 437 nm at a concentration of 0.5 mg/mL, whereas the Ch/P1 system at the same concentration gives NPs with an average diameter of 276 nm. The distribution of the substituted cycles along the macromolecular chains in the conjugates would explain these differences.
Apparent hydrodynamic diameter by intensity (Dh), polydispersity index (PDI), and zeta potential (ζ) values of Ch/P NP and RP-loaded Ch/P NP.
RP: retinyl palmitate; NP: nanoparticle; Ch/P: palmitoyl chitosan.
2 mM NaCl solution.
When RP was encapsulated into the NPs, Dh values of RP-loaded Ch/P1 NP did not significantly change and those of RP-loaded Ch/P2 NP reduced by 50%, indicating again a different behavior by the derivatives in the presence of RP. However, in this case, the hydrophobic interactions between the Ch/P2 amphiphile and the antioxidant produce a more compact and constrained core. It can be concluded that the relatively high LE values, and the considerable decrease in the Dh values of RP-loaded Ch/P2 NPs indicate a stronger affinity for the antioxidant derivative by the amphiphilic chitosan obtained by Procedure 2.
The zeta potential (ζ) of the NPs was measured in a saline solution with an ionic strength (Ic) higher than 10−3 M. 42 Positive values (Table 2) were obtained for all systems, confirming that the NPs were composed of hydrophilic chitosan outer shells and hydrophobic cores formed by palmitoyl moieties and the RP antioxidant for loaded NPs, as reported for other systems.51,52 All ζ values were higher than 20 mV, indicating that the NP suspensions are stable in aqueous medium 53 due to the positively charged amino groups of the hydrophilic chitosan polymer at the NP surface. The ζ values of RP-loaded NPs were somewhat higher (around 30 mV) than those of blank NPs (around 25 mV), indicating that the RP molecules are located in the core of the NP and also that the entrapment of RP contributes to the stability of the NPs. Similar ζ values were reported for chitosan NPs loaded with vitamin E or mixtures of vitamin E and vitamin C. 54
An examination of SEM images (Figure 3) of the NPs revealed an almost spherical morphology for all blank and RP-loaded NPs. It was confirmed that the unloaded Ch/P2 NPs had the largest size as determined by DLS. The Ch/P2 system was further examined by TEM (Figure 4) and again a pseudospherical morphology was observed in both loaded and unloaded NP. The typical core-shell structure was slightly more pronounced for RP-loaded NPs compared with the unloaded NPs, presumably because of the intensification of the interactions in hydrophobic microdomains. It must be pointed out that in the TEM images, the self-aggregated NPs had smaller sizes than those observed by DLS, which was attributed to the sample being in a dried state, 50 whereas DLS determines the size in the hydrated state. A schematic presentation of the NP with the palmitoyl moiety and RP is presented in Scheme 1.

SEM images of blank and RP-loaded NP.

TEM images of blank and RP-loaded NP for the amphiphile obtained by Procedure 2.
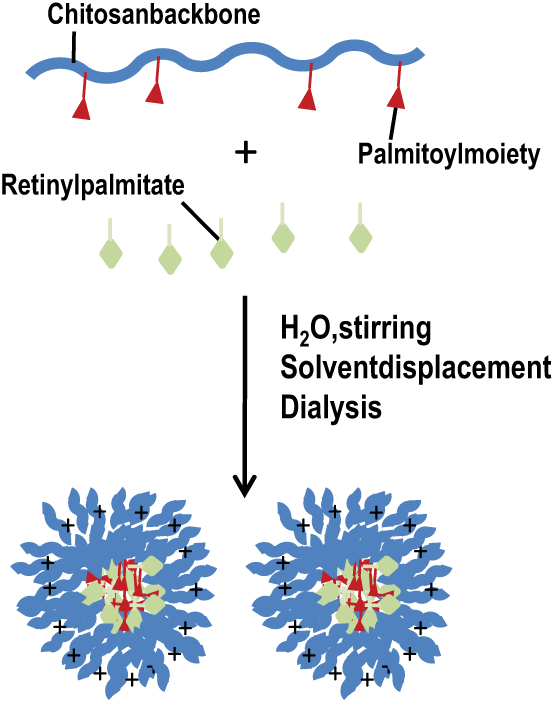
Representation of the formation of palmitoyl chitosan (Ch/P) conjugates nanoparticles charged with retinyl palmitate (RP) and stabilized by hydrophobic interactions between the palmitoyl (P) and RP moieties.
Thermal characterization
The main parameters obtained from TGA and derivative thermogravimetry (DTG) (first derivative of the TGA curve with respect to temperature) of the conjugates and pristine compounds are shown in Table 3. An increase in the value of T5% was observed in the samples of both the amphiphiles and the RP-loaded NPs when compared to chitosan. The weight loss produced at this initial stage is mainly associated with the absorption of water in the hygroscopic chitosan, and the shift of T5% up to 60°C–70°C for the amphiphilic derivatives and up to 100°C–150°C for RP-loaded NPs can be correlated with an increase in hydrophobicity due to the incorporation of palmitoyl residues and the hydrophobic antioxidant in the corresponding NPs.
TGA and DTG results of starting materials, Ch/P conjugates, and RP-loaded Ch/P NP.
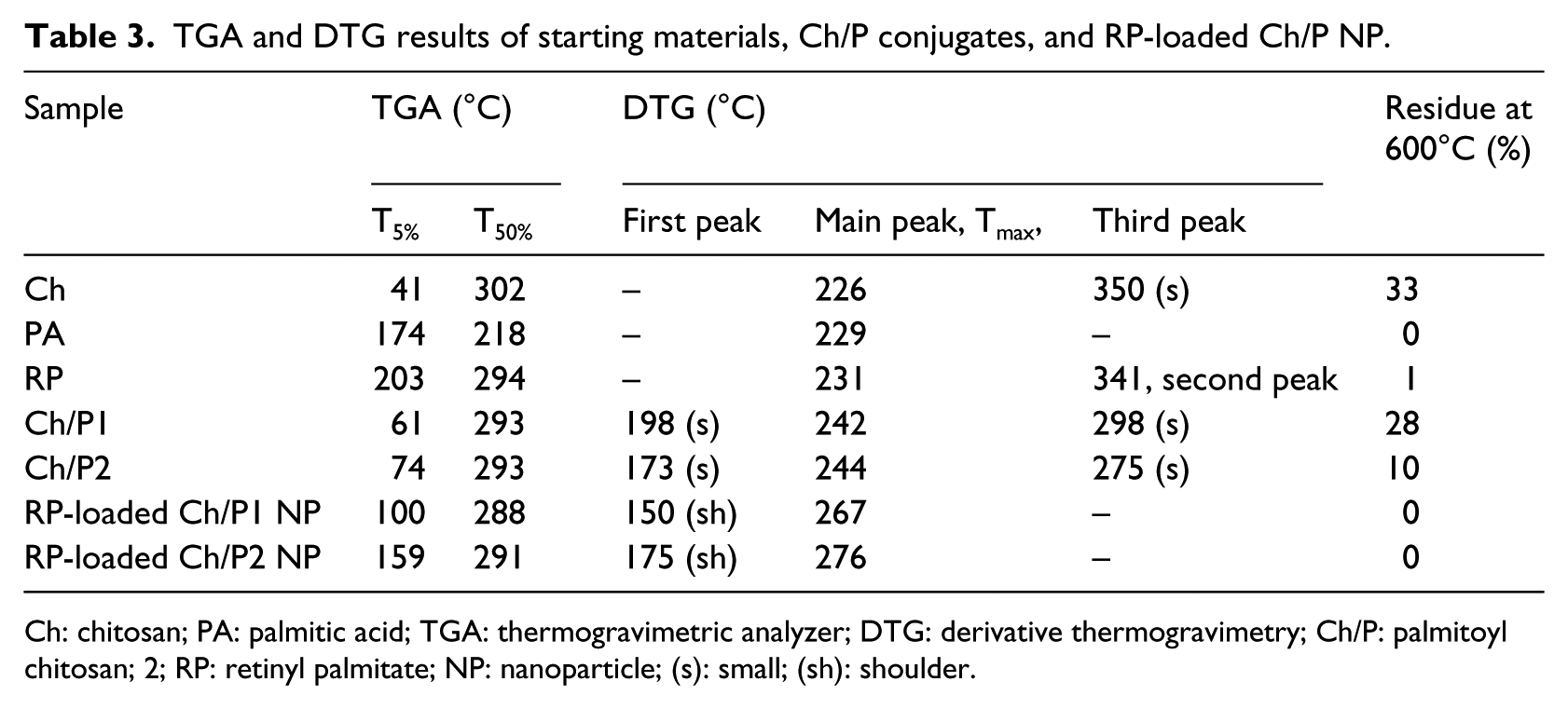
Ch: chitosan; PA: palmitic acid; TGA: thermogravimetric analyzer; DTG: derivative thermogravimetry; Ch/P: palmitoyl chitosan; 2; RP: retinyl palmitate; NP: nanoparticle; (s): small; (sh): shoulder.
For Ch/P samples, thermal degradation occurred between 100°C and 400°C. Different levels of weight loss were observed that reflected the degradation of the different components of the materials. Small maxima in the DTG curves of Ch/P1 and Ch/P2 at 198°C and 173°C, respectively, were attributed to the degradation of palmitoyl moieties. 48 The main maxima of the main degradation stage at 242°C and 244°C for Ch/P1 and Ch/P2, respectively, were ascribed to deacetylation of chitosan and the cleavage of glycosidic linkages. Afterward, the complete decomposition of chitosan involving pyrolitic processes was reflected in a small and broad peak around 300°C.55,56 These data confirmed the coupling reaction.
For the RP-loaded Ch/P NP, thermal degradation also took place between 100°C and 400°C. The DTG curves presented undefined shoulders around 150°C and 175°C for RP-loaded Ch/P1 and Ch/P2 NP, respectively, that were associated with the degradation of palmitoyl residues, followed by broad maxima at 267°C and 276°C, respectively. This main degradation stage includes the bulk degradation of chitosan and RP. On the other hand, the signal that appeared at 600°C in the Ch and Ch/P samples disappeared in the RP-loaded NP samples, which could suggest a change in hydrogen bond interactions between the polysaccharide skeletons 57 due to the NP formation in conjunction with the encapsulation of the antioxidant. This is a clear demonstration of the interactions established in the NP between the RP molecules and the palmitoyl moieties.
In vitro biological assays
One of the main functions of retinol in skin is to neutralize unstable oxygen molecules and participate in the process of collagen synthesis. However, the sensitivity of this molecule to light is well recognized, along with its strong hydrophobic character. The preparation of self-assembled NPs provides a safer manipulation and application of this compound. The antioxidant activity of the RP-loaded NPs was evaluated by an in vitro DPPH• assay.58,59 This assay is frequently used to assess the antioxidant potential of vitamins 54 and has been applied to different chitosan NPs that encapsulate 33 or conjugate60,61 antioxidants. In this work, the free RSA of the RP was studied, and the reaction kinetics were obtained when an ethanolic solution of DPPH• (0.127 nm) was incubated with different RP concentrations. The results are shown in Figure 5. For this antioxidant, the steady state was reached at 350 min. This type of kinetic reaction is considered to be slow, 59 as reported for other antioxidants such as guaiacol. 62
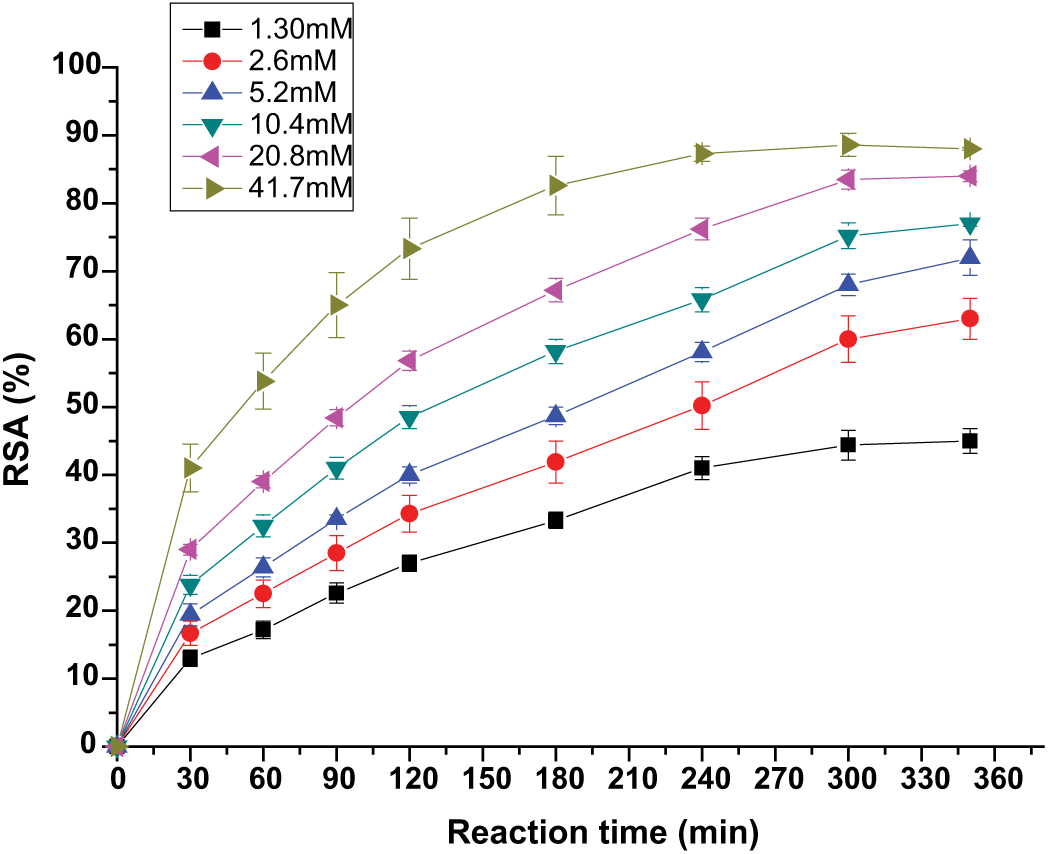
Reaction kinetics of RP in a DPPH• assay. Data represent mean ± SD (n = 5).
For the evaluation of the antioxidant activity of blank and RP-loaded NPs, ethanol was selected as release medium since it is a good solvent for RP. Then, the RSA of NP extracts taken after 48 h of release in the dark was monitored over time. The results are shown in Figure 6. In this case, the stationary state was reached after 30 min. This shorter reaction time was due to the lower concentration of RP present in the extracts. The RSA values of RP-loaded Ch/P1 NPs and RP-loaded Ch/P2 were 33% ± 2.3% and 39% ± 3.3%, respectively. Extracts of blank NPs taken under the same conditions presented a similar behavior; they had an RSA of 25% ± 3.2% after 30 min. For this sample, a slight decrease in RSA was observed over time. This decrease has been related to the reversibility of the reaction between the antioxidant and DPPH in some cases. 63 The fact that blank NP presented RSA confirms the antiradical properties of the polysaccharide, which has been ascribed to the amino groups at the C-2 position. 64 In addition, it was recently reported that this property is enhanced when Ch forms NPs. 30
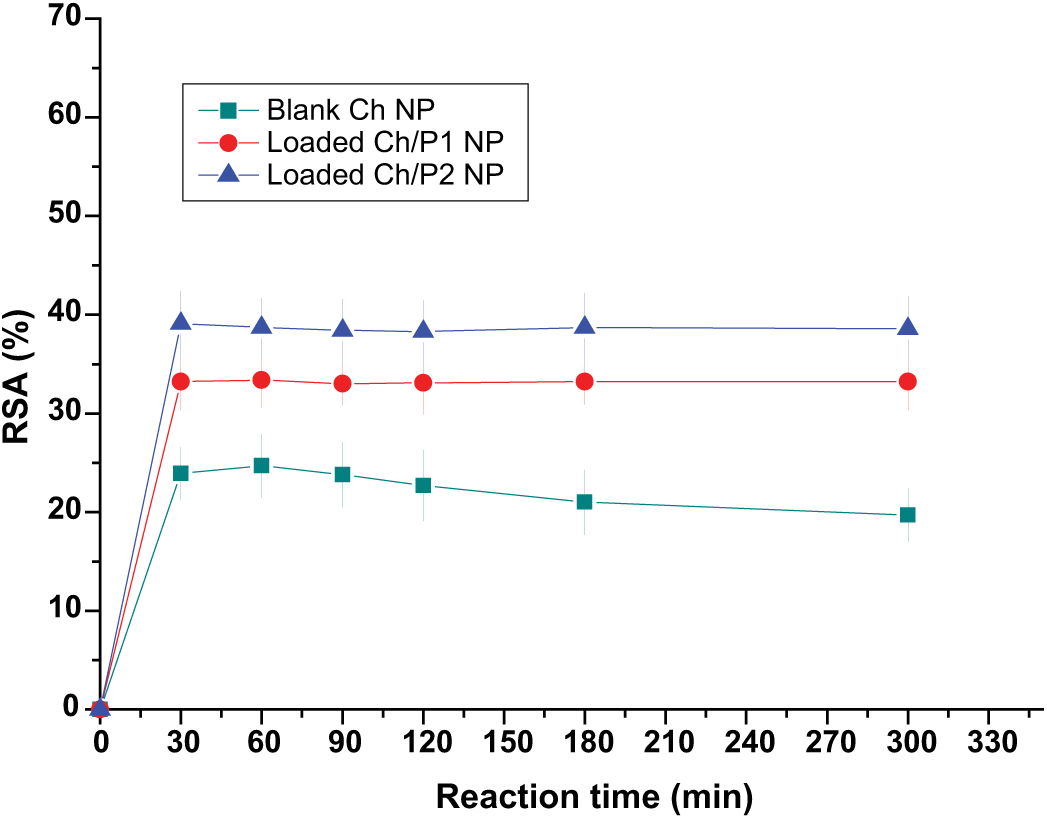
Experimental RSA of DPPH with time in the presence of ethanolic extracts of blank and RP-loaded NP. Data represent mean ± SD (n = 5).
Biocompatibility is a major concern in biomaterials science. Thus, the biocompatibility of the RP-loaded Ch/P2 NPs was tested in HFB using an MTT assay. This test is dependent on the activity of a mitochondrial enzyme, succinate dehydrogenase, which may be impaired after cells are exposed to toxic species. 65 For the assessment of cytotoxicity, the cells were incubated with dispersions of blank and RP-loaded NPs at different concentrations and dosages. The results are shown in Figure 7. Blank NPs exhibited minimal cytotoxicity, which was independent of the dose. The cell viability (CV) was around 100%. No significant reduction in relative CV for RP-Ch/P2 NPs concentrations lower than 15.9 µg/mL was detected, but they exhibited significant reductions (<20%) at concentrations between 15.9 and 250 µg/mL. However, at all concentrations the CV was higher than the limit indicated by the ISO standard for biocompatibility. 66 It seems that the mitochondrial enzyme activity and proliferation of fibroblasts were not affected by blank and RP-loaded NPs at concentrations lower or equal to 250 µg/mL. These results correlate well with those reported by Errico et al. 4 for RP-loaded poly(lactic-co-glycolic acid) (PLGA) NPs on HaCaT and 3T3 cell lines.
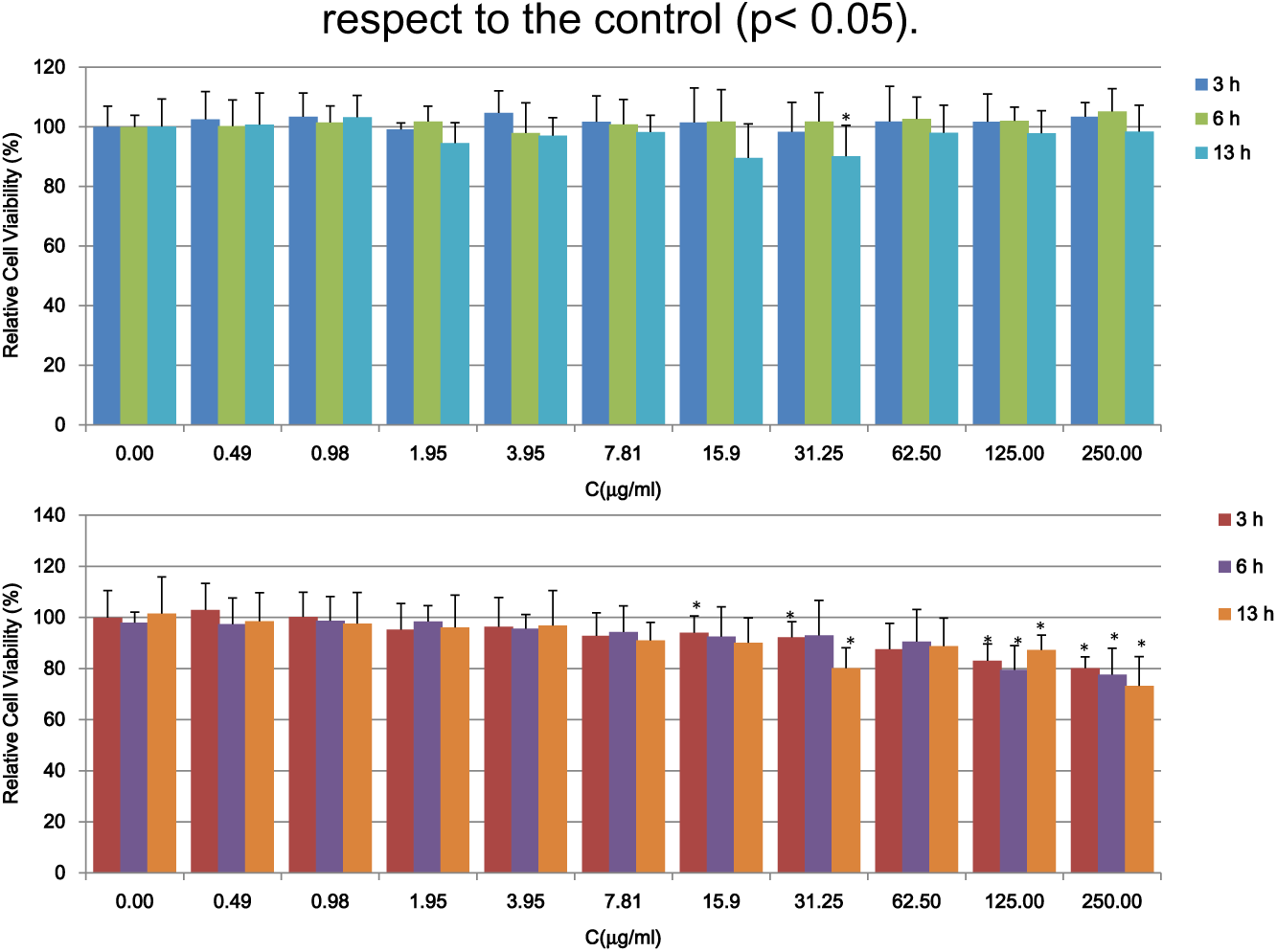
MTT results of blank (upper) and RP-loaded Ch/P2 NPs (lower). All results are shown as mean ± SD (n = 16). Asterisk (*) depicts a significant difference between results of the corresponding sample with respect to the control (p < 0.05).
The LDH assay was used to evaluate whether RP-loaded NPs exhibited any membrane-disruptive effects. This test measures cell lysis through the LDH activity, which is present upon cell death. 67 The relative LDH release in both blank and RP-loaded Ch/P2 NP samples was zero at concentrations lower or equal to 250 µM, clearly indicating the absence of membrane toxicity. The obtained results were consistent with those obtained by the MTT assay.
The cellular uptake of RP-loaded Ch/P2 NPs by fibroblasts with incubation times of 6 and 18 h was visualized by fluorescence microscopy. The effect of incubation time is clearly shown in Figure 8. After 6-h incubation, only a few NPs were observed inside the cells, and they were located in the periphery. With increasing incubation time, more NPs were taken up by the cells, and they were located close to the nuclei.
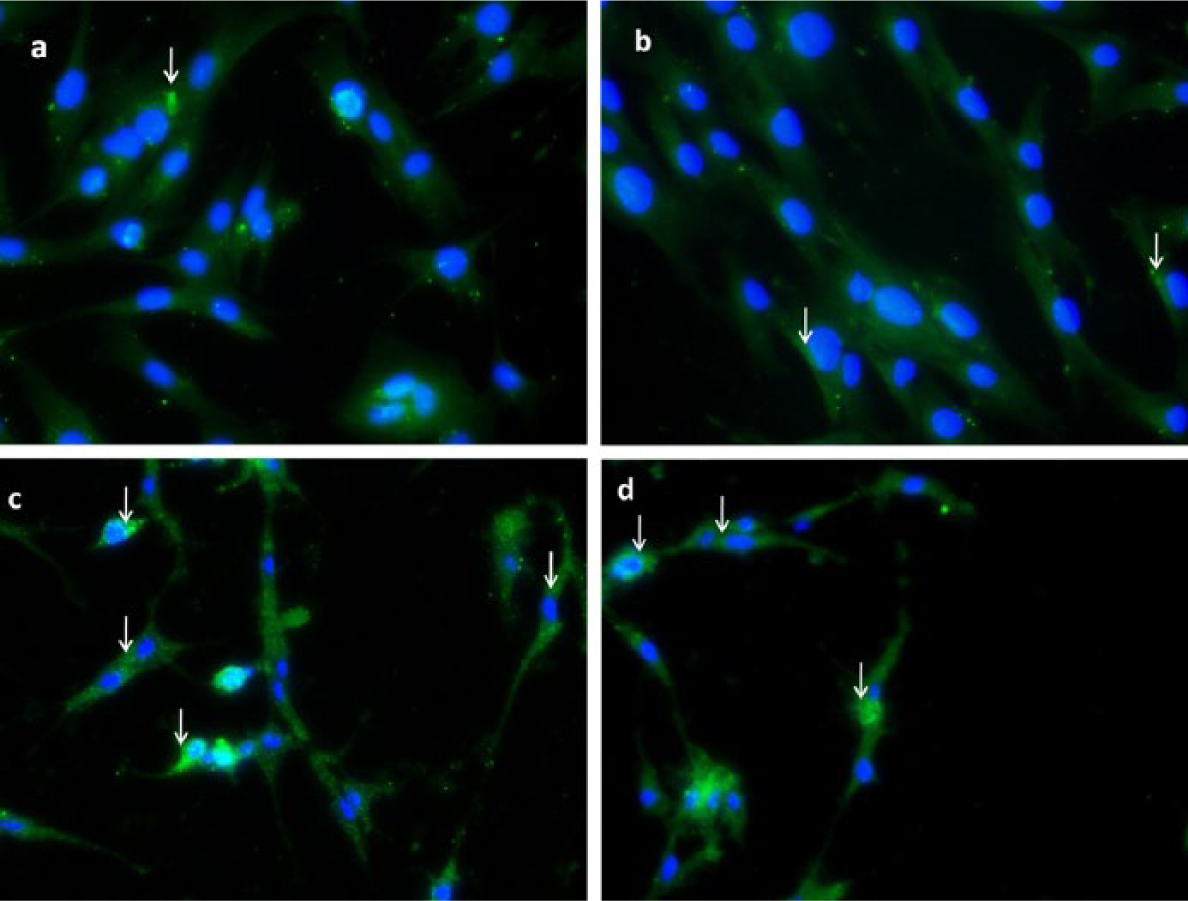
Fluorescence microscopic images of human fibroblasts incubated with RP-loaded Ch/P2 NP at 50 µg/mL: (a) after 6-h incubation (20×), (b) after 18-h incubation (20×), (c) after 18-h incubation (10×), and (d) after 18-h incubation (10×).
The in vitro data have promising potential for this nanocarrier system for application in dermal tissue, and a more in-depth study of its biological properties will be the objective of future work.
Conclusion
An amphiphilic and self-assembling chitosan based on a Ch/P conjugate was successfully synthetized and self-assembled into NPs in aqueous medium and RP was encapsulated at a high loading efficiency. The synthesis of the Ch/P conjugate mediated by the EDC/NHS coupling agent at mild conditions is proposed as a convenient method. The RP-loaded Ch/P NPs have a pseudospherical morphology with a typical core-shell structure, a mean NP diameter of around 280 nm and a zeta potential of 29 mV, which assures stability in aqueous medium. In vitro, the RP-loaded NPs had antioxidant activity which was attributed to the contribution of both chitosan and RP with an apparent RSA of about 40%. Cell cultures of the RP-loaded NPs had minimal cytotoxicity and membrane-disruption toxicity along with a high uptake in HFB. Overall, this system, based on a chitosan conjugate that possesses the same hydrophobic moieties as the loaded retinoid, is a promising nanocarrier with antioxidant properties for potential applications in dermal tissue. Additionally, the concept proposed here could be expanded to other amphiphile/antioxidant systems.
Footnotes
Acknowledgements
The authors thank CIBER-BBN for financial support and R.A. Ramírez for the support with cell culture assays.
Declaration of conflicting interests
The authors declare that there is no conflict of interest.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.