
Abstract
Filler materials of soft tissue should have good biodegradable absorption and biocompatibility. To investigate feasibility of keratin hydrogels used as filler materials, a keratin with high molecular weight and self-assembly ability was extracted from human hair using a modified reduction method and was characterized using sodium dodecyl sulfate–polyacrylamide gel electrophoresis, Fourier transform infrared, and X-ray diffraction. An injectable keratin hydrogel was also prepared and its water absorption, cell toxicity, and histological behavior were evaluated by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide method and subcutaneously implanted experiment. The results showed that the modified extraction method could keep the activity of the keratin and provided the self-assembly crosslinked ability of the keratin. The water absorption of the keratin hydrogel could be controlled by adjusting some preparation parameters and the percentage of water absorption was up to 850%. In addition, the subcutaneous injection experiment for Sprague Dawley rats indicated that the keratin hydrogel had good biocompatibility and could promote the formation of angiogenesis as well as proliferation. It can be predicted that the keratin extracted from human hair and keratin hydrogel have good application prospects in the field of filler materials for soft tissue.
Introduction
Keratin has become one of the most important biomaterials among commercially available biomacromolecules due to its good formatting-film ability, high physicochemical and excellent mechanical properties, as well as relatively good biocompatibility.1–4 Aside from hair keratins, other keratin group members include epithelial and hair follicle–specific epithelial keratins. Hair keratins have relatively high sulfur content in their head and tail globular domains and are further reinforced by a continuous phase of highly crosslinked matrix proteins referred to as keratin-associated proteins.4–6 Until now, various structural forms of extracted keratins together with some chemical modifications 7 or combinations8–10 have been prepared for biomedical purposes, including films,2,11,12 hydrogels, 13 scaffolds,14–16 bone regeneration, 17 hemostasis, 18 and peripheral nerve repair.19,20 However, most of the publications primarily focused on extraction, physicochemical, and cellular aspects of the prepared keratin biomaterials. Those studies with human hair keratin (HHK) and keratin hydrogel used as filler materials of soft tissue were really few.
The keratin proteins extracted from hair can be classified into three broad groups: alpha-, beta-, and gamma-keratins. 21 α-Keratins are an alpha-helical tertiary structural proteins and have an average molar mass in the range of 60–80 kDa. γ-Keratins are globular and low in molecular weight. However, β-keratins are primarily protective and form the majority of the cuticle. 22 So β-keratins are difficult to extract and lower in molecular weight than the other types of keratins. Numerous methods for oxidation and reduction have been established and published for the extraction and purification of keratins from hair.23,24 These methods rely on chemical processes to break down the extensive disulfide crosslinked network of the fiber, combined with or followed by an extraction step in which the proteins are denatured and solubilized. However, some chemical crosslinked agents are used to promote the crosslinking of keratins and form hydrogels.
The aim of this study is to produce a HHK with high molecular weight using a modified reduction method and to prepare an injectable keratin hydrogel. In addition, this study also focuses on the biological evaluation of the keratin hydrogel. Water absorption, cell toxicity, and histological behavior of the keratin hydrogel will be evaluated by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) method and subcutaneously implanted experiment in vivo. These relevant studies are expected to provide important data for soft tissue applications of keratinous biomaterials.
Materials and methods
Materials
Human hair was obtained from a local barber. Urea, 2-mercaptoethanol (Shanghai Beier Chemical Ltd, China), thiourea, ethanol, trichloromethane, ammonium hydroxide, hydrogen peroxide, sodium dodecyl sulfate (SDS), and other chemicals of analytical grade were commercially available and were purchased from Guangzhou Chemical Reagent Factory (China). Tris–HCl was purchased from Amresco (USA). SDS–polyacrylamide gel electrophoresis (PAGE) was purchased from BL502A (Biosharp, China).
Extraction of HHK
The prepared method of the keratin extraction from human hair fibers was modified according to the reported methods with some modifications.16,25 Briefly, human hair from a local barber shop was washed with alkaline detergent for three times, and external lipids were removed with a mixture of chloroform/methanol (2:1, v/v) under reflux at 70°C for 2 h. The clear, dry, and delipidized hair was decolorized using a mixture of hydrogen peroxide and ammonia (2:1, v/v). The decolorized hair was refluxed with a mixture of urea (45 wt%), SDS (4 wt%), and mercaptoethanol (3 wt%) at 55°C for 12 h. After the mixture was filtered and centrifuged at 4000× g for 20 min at room temperature, the obtained supernatant was dialyzed against deionized water using dialysis bag (molecular weight cutoff of about 10 kDa) and the outer water was replaced with distilled water four times a day. The dialysis was rapidly concentrated in the dialysis bag embedded into water absorption polyacrylic acid resin, and then the concentrated solution was diluted to form 10 wt% keratin solution and the extracted keratins were kept sealed dry preservation.
Fabrication of the injectable keratin hydrogel
The extracted keratin solution placed using solution casting method and through oxidation crosslinking for oxygen was made to form keratin films at room temperature for 24 h. The keratin films were milled and filtered using 200 mesh. Finally, the proper keratin powders were loaded into a syringe which had been sterilized with 75% ethanol and washed with saline for three times. The keratin powders in the syringe showed gel particles and could be used easily to inject into soft tissue.
SDS–PAGE analysis
The extracted keratins were performed using SDS–PAGE and using MiniProtein II vertical electrophoresis system (MiniProtein II; Bio-Rad Co., China). The above keratin solution was first diluted to 1.0 mg/mL with ultrapure water, and then the diluted solution (100 µL) was mixed with 100 µL of SDS sample buffer. The mixture solution was boiled for 5 min to denature the keratin and immediately placed on the ice. The denaturing solution (20 µL) was loaded onto 5%–12% gradient Tris–HCl gels. Separation was carried out at 80 v for 30 min, followed by 120 v for 100 min. After electrophoretic separation, the gels were rinsed with purified water for 5 min and then stained with 0.1% Coomassie Brilliant Blue R-250 for 2 h, followed by destaining with 10% acetic acid and 40% ethanol. Finally, the treated keratin samples were compared to a standard sample of protein.
Structural and morphology analysis
The freeze-drying keratin, human hair, and HHK were tableted with KBr and characterized using Fourier transform infrared (FTIR) spectroscopy (TENSOR 27; Bruker, USA), respectively.
The fracture surface of the HHK and the keratin hydrogels after freeze-drying were characterized using field emission scanning electron microscopy (Ultra 55 FE-SEM; Zeiss, Germany).
Wide-angle diffraction analysis
The freeze-drying keratin, human hair, and HHK were milled and filtered using 200 mesh, respectively. These samples were characterized using wide-angle diffraction (WAXD; MSAL-XD 2 automatic X-ray diffractometer, China) to measure crystallization properties. Testing conditions were as follows: Cu Kα radiation in the range of 2θ = 10°–45°, a scanning rate of 4°/min, 36 kV of voltage value, 20 mA of current value, measuring range between 10° and 45°, and 0.01° of scan step size.
Water absorption
Water absorption of the keratin film can indirectly reflect hydration properties of the keratin hydrogel. The above keratin films were divided into two groups. Group 1 was dried under vacuum at 40 °C and Group 2 was not dried. The two group samples weighted as S were immersed in distilled water and maintained at 25°C ± 2°C over 2 h, during which time they were removed from the water at 20-min intervals (e.g. 20, 40, 60, 80, 100, and 120 min), gently blotted with tissue paper to remove excess water from the surfaces, immediately weighed to the nearest 0.001 g four times, and then returned to the water. The average of the masses measured at each 20-min interval was calculated and these average masses were designated Swet. Water absorption (S%) was determined according to the following equation and an average of four values was taken as per equation given to
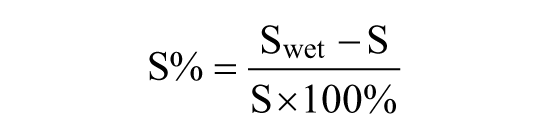
where S represents the weight of the keratin film after drying.
MTT
MTT assays were used to evaluate quantitatively cell proliferation and viability as well as toxicity in the presence of keratin. L929 murine fibroblasts (provided by Southern Medical University, China) were cultured in culture flasks with Dulbecco’s modified Eagle’s medium (DMEM) (provided by Gibco-BRL company, USA)-containing serum (provided by Hangzhou Evergreen Biological Engineering Co., Ltd, China). The gels were extracted according to ISO standard 10993-5. The 2 g gels were extracted with pure DMEM medium for 24 h. The negative control consisted of pure DMEM medium and the positive control consisted of 5% dimethylsulfoxide (DMSO) solution.
Based on the extract solution and bovine serum as culture medium, cell viability was examined using optical microscope at a screened culture interval (12, 24, 48, and 72 h) and the cells were cultivated at 37°C in a humidified incubator in a 5% CO2 atmosphere. Each cell-cultured well plate was injected with 20 µL MTT solution and placed in a dark environment. After further culturing for another 4 h, the supernatant was removed and discarded carefully, and then 150 µL DMSO was added into each well plates to dissolve the formazan. The well plates were shaken and stirred with a vortex mixer for 10 min to ensure a uniform solution. Finally, the absorbance of the solutions in a 96-well plate was recorded using a microplate reader at 570 nm using ultraviolet (UV) spectrophotometer and OD570 values were obtained. Relative cell proliferation rate (RCV%) was calculated from the following formula
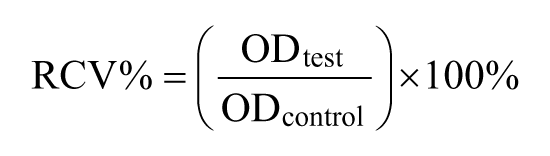
Subcutaneous implantation
Six Sprague Dawley (SD) rats (provided by Experimental Animal Center of Guangdong Province) were anesthetized and injected using 0.3 g of keratin hydrogel into the subcutaneous tissues. At selected implantation intervals (e.g. 4 and 7 days), the surrounding tissues (the size is 2 × 1.5 × 0.5 cm) of the injected keratin hydrogels were fixed and stained for histological observations.
Results and discussion
Extraction process of the keratin and fabrication of the keratin hydrogel
To improve the average molecular weight of the HHK, a modified reduction method was used during keratin extraction. First, the human hair was partly dissolved and matrix proteins were removed which are surfaced on the hair using the oxidation reaction. And then the oxidized human hair was extracted using the reduction reaction. In the extract process, the β-keratin content of the HHK was decreased and its average molecular weight was improved. In addition, the oxidation reaction and the decoloring process were simultaneously carried out using the mixture solution with hydrogen peroxide and ammonia to dissolve partly the β-keratin. For the reduction reaction, 2-mercaptoethanol was used as reductant, urea was used as denaturant, and SDS solution was used as stabilizer.
In the work, the extraction method of keratin from human hair was improved according to the reported methods.16,25 During extraction and enrichment of the human hair, lye was not used to adjust pH of extract solution. The extraction method can not only avoid the reduction reaction between lye and thiol of keratin molecule but also retain the activity of thiol and self-assemble ability of the keratin. The crosslinking reaction without crosslinking agent could be carried out between the molecules in air in order to improve the mechanical properties and degradation of the keratin hydrogel. To reduce intermolecular crosslinking and hydrophobic aggregation resulting in forming precipitation during keratin extraction, the super absorbent resin was used to not only enrich rapidly the keratin solution but also promote formation of reticular structure and preparation of keratin hydrogel.
SDS–PAGE analysis
Analysis of the molecular weight of keratin samples showed a characteristic banding pattern. Figure 1 is the SDS–PAGE figure of the extracted keratin, the two middle bandings are keratin solution samples, and the left and right bandings are the standard protein. In contrast, the three bandings of the extracted keratin correspond to characteristic bands of the standard protein at 30, 50, and 65 kD, of which the distribution at 65 kD is narrow but the distribution at 30 and 50 kD is wide and the color at 50 kD is deep.
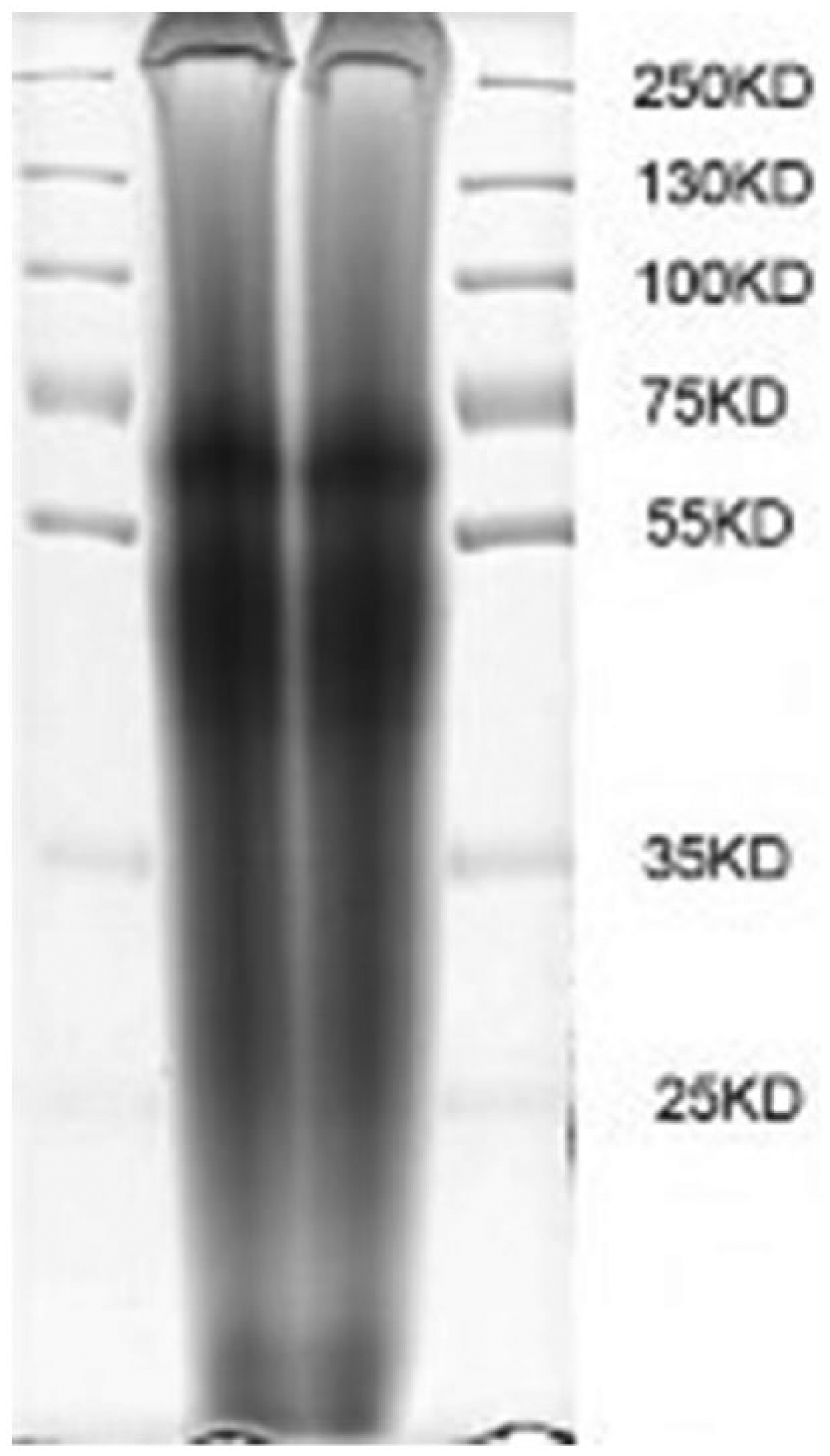
SDS–PAGE of the extracted keratin solution. Two middle bandings are keratin solution samples; left and right bandings are standard protein.
Structural analysis
FTIR spectrum of the human hair, freeze-drying keratin, and HHK are shown in Figure 2. Characteristic absorption bands of peptide bonds (–CONH–) for human hair and HHK were observed. The vibrations of the peptide bonds can be attributed to amide A, amides I, II, and III. The adsorptions at 3300 cm−1 arised from the N–H stretching vibrations of amide A. Amide I band due to C=O stretching vibration occurs in the range of 1700–1600 cm−1. Sharp peak was also observed at 1656 cm−1. Amide II, which falls at 1541 cm−1, is related to N–H bending and C–H stretching vibration. Amide III band (1220–1300 cm−1) was observed at 1233 cm−1 as a sharp peak. The strong absorption peak centered at 625 cm−1 can be attributed to the C–S band stretching vibrations, validating the presence of reduced (–SH) form of keratins. 26
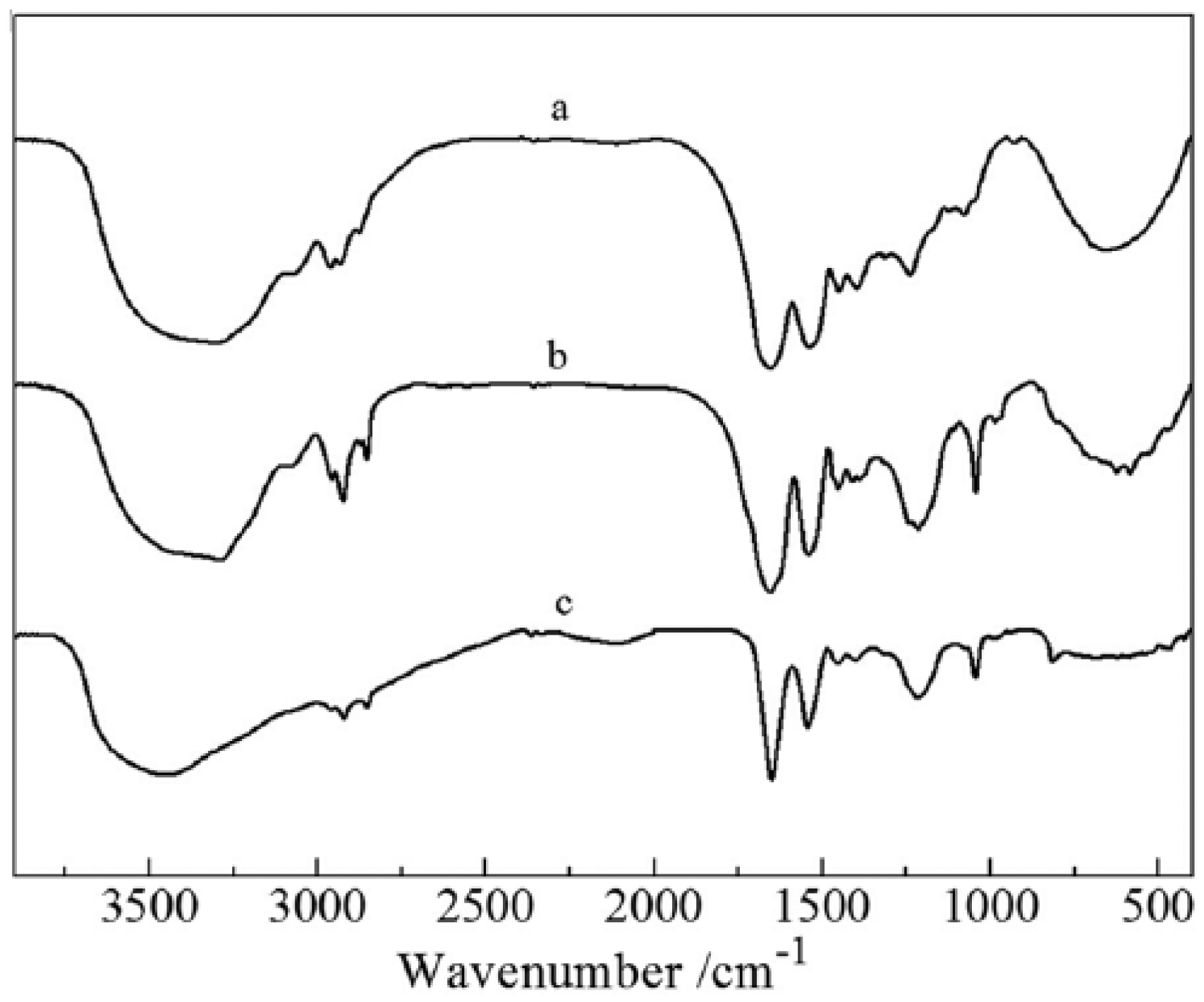
FTIR spectrum of the human hair, freeze-drying keratin, and human hair keratin.
Compared to the FTIR spectrum of human hair, the characteristic peaks of amides II and III for HHK changed very small. However, the characteristic peaks at 2876 and 2962 cm−1 for human hair shifted at 2962 and 2942 cm−1 for HHK. This may be because the disulfide bonds (–CH2–S–S–CH2–) are broken during extracted process and the electron density of C–H is reduced, and then C–H stretching vibration shifted to higher. In addition, the C–S stretching vibration at 985 cm−1 and C=S stretching vibration at 1043 cm−1 for the extracted keratin occurred, suggesting that part of disulfide bonds for human hair converted into sulfonic acid groups. Therefore, the keratin solution extracted from human hair still possesses some features of polypeptide.
WAXD analysis
To further determine structural changes in keratin films during preparation process, the human hair, freeze-drying keratins, and HHK were characterized using WAXD and shown in Figure 3. It could be seen from the figure that a slight peak for human hair occurred at 20.33°, a sharp and disrupt peak for the freeze-drying keratin appeared at 20.54°, but a clear and sharp peak for HHK generated at 20.88°.
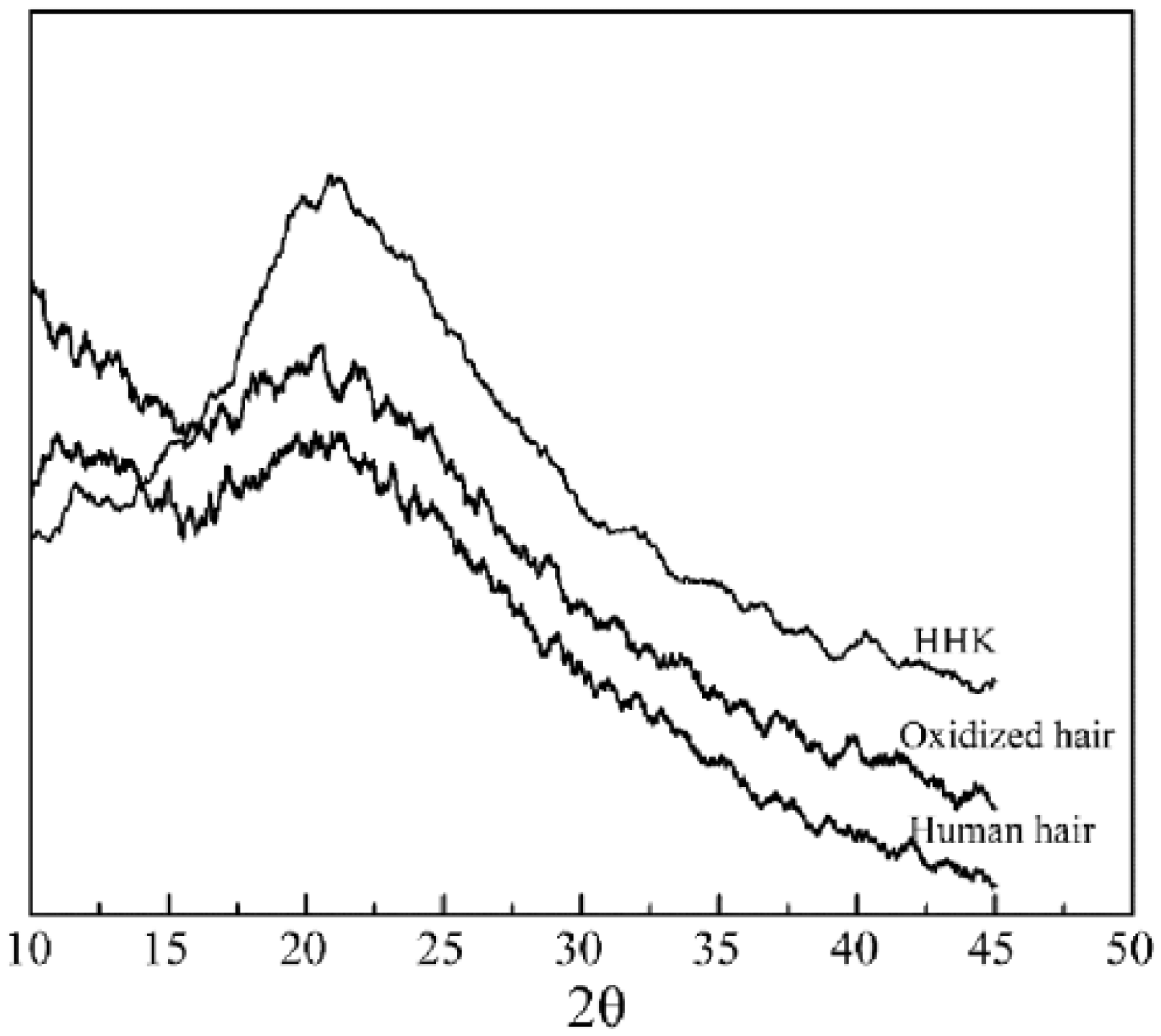
WAXD of the human hair, freeze-drying keratin, and human hair keratin. A slight peak for human hair at 20.33°, a sharp and disrupt peak for the freeze-drying keratin at 20.54°, and a clear and sharp peak for HHK at 20.88°.
The diffraction peaks of the human hair and HHK were between 20° and 21° and were consistent with some literatures, indicating that the keratins during the extracted process could retain the β-keratin helix structure of human hair.2,27 The obvious signal for the keratin films suggested the keratin molecules produced well-ordered array during forming films. The results confirmed further that the keratin molecules produced a disulfide bond restructuring and crosslinking during forming films of the keratin solution. 28 For the freeze-dried keratin, many sharp peaks occurred due to the influence of some unremoved hydrolysis and degradation products during dialysis. Compared with the human hair and the keratin films, the diffraction peak area of the freeze-dried keratins was not obvious and structural orderly degree of the freeze-dried keratins was lower.
Water absorption
Water absorption of the keratin films was measured to reflect water absorption of the injectable hydrogel and is shown in Figure 4. The maximum water absorption of the keratin film with drying was only 320% (Figure 4 (A)); however, those of the keratin film without drying was up to 850% (Figure 4 (B)) and its moisture content was 142%. Additionally, the change curves A1, A2, B1, and B2 in Figure 4 were water absorption for the keratin films which are, respectively, dried and not dried. The water absorption and maximum water absorption for the keratin films with drying were close.
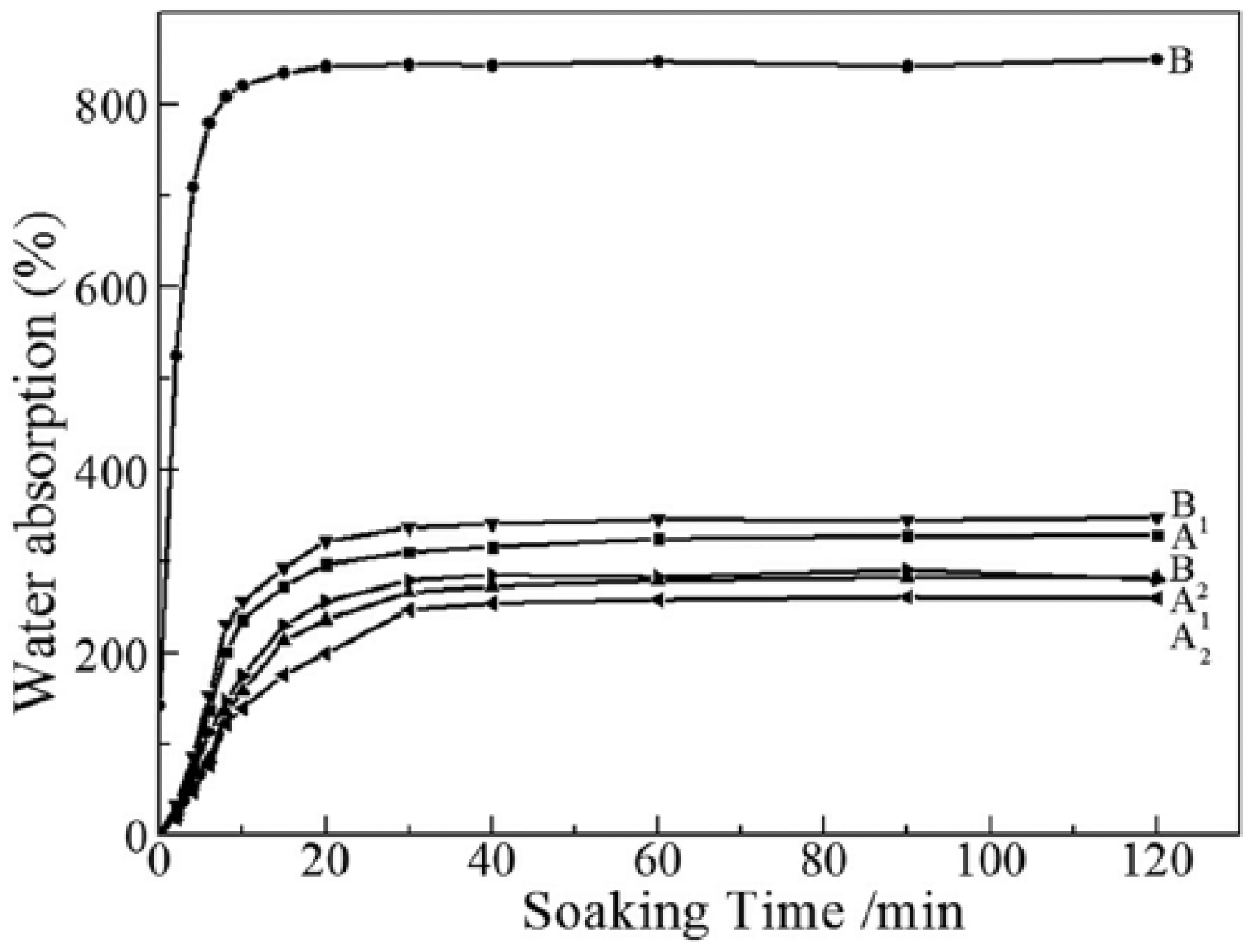
Water absorption of the keratin films (A: with drying; B: without drying). The water absorption of the keratin film without drying was up to 850%.
The experimental results can provide a new method to control water absorption of the keratin hydrogel. The keratin film or powder without drying can keep high water absorption or swelling. The method that controls drying degree can adjust water absorption and will provide more application for keratin hydrogels. In addition, the soaking time up to the maximum water absorption for the keratin film with drying was 20–30 min, but the soaking time for the keratin film without drying was only 10 min, suggesting that the swelling rate for the keratin film without drying was higher than that of the keratin film with drying. Some literatures have reported that water absorption of keratin sponges is between 367% and 554% and the time up to the maximum water absorption exceeds 20 h. 29 Interestingly, according to our work, the prepared keratin hydrogels have higher water absorption and shorter soaking time up to the maximum water absorption. The scanning electron microscopy (SEM) image of the fracture surface for the HHK and the keratin hydrogels after freeze-drying is shown in Figure 5. The spongy and lamellar structure of the HHK and keratin hydrogels was beneficial to water absorption.
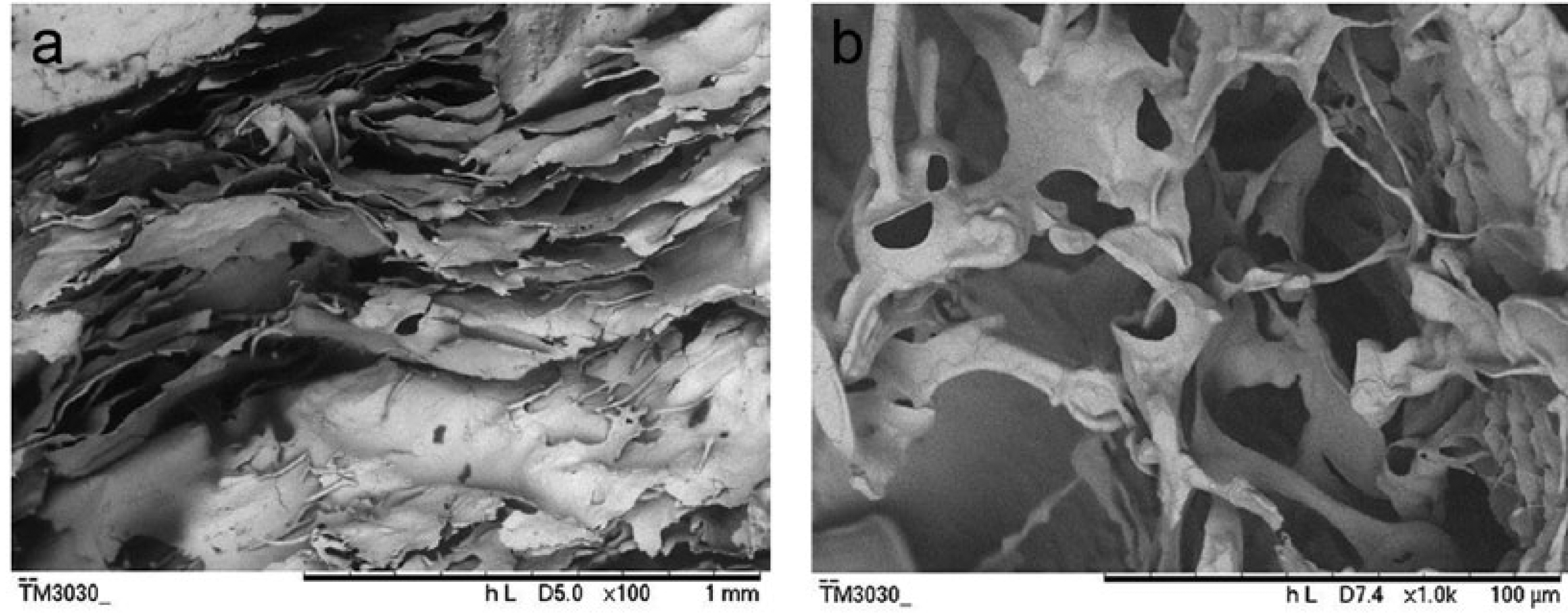
SEM image of the fracture surface for the human hair keratin and the keratin hydrogels: (a) the human hair keratin after freeze-drying and (b) the keratin hydrogels after freeze-drying.
MTT
Cell growth and attachment of L929 murine fibroblasts for blank control, keratin hydrogel, and positive control after different culture intervals are shown in Figure 6. Cell numbers under blank control and extracted keratin solution were increasing with culture time. In the meantime, cell morphology was normal. Only a small number of dead cells were circular and in agreement with normal cell metabolism. However, cells under positive control were almost dead and appeared circular as well as few cell debris. Based on OD570 value calculation, RCV% of the extracted keratin solution for 24, 48, and 72 h culture time was 119%, 122%, and 123%, respectively.
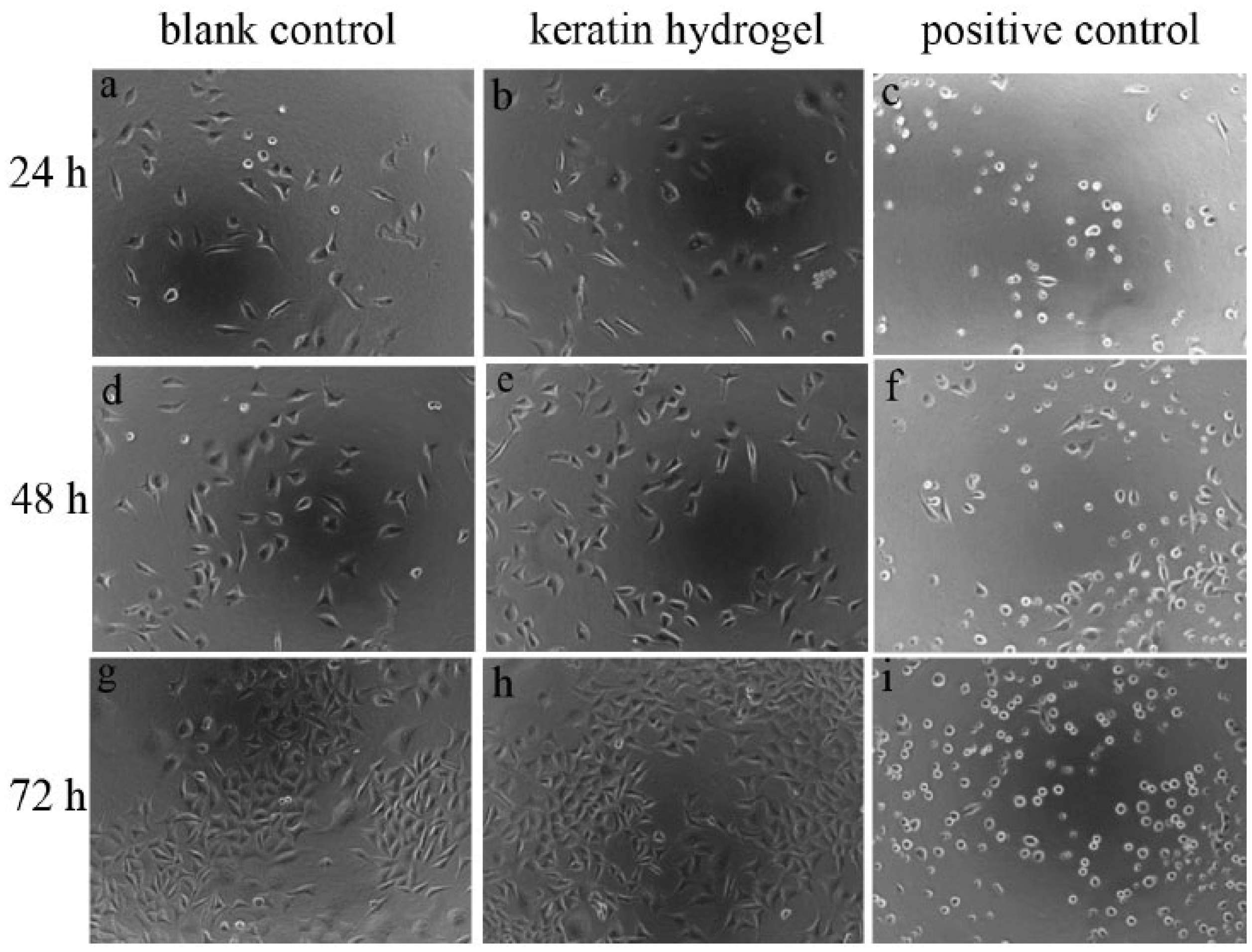
Confocal micrographs of cell growth and attachment for blank control (a, d, g), keratin hydrogel (b, e, h), and positive control (c, f, i). L929 murine fibroblasts were cultured in culture flasks with DMEM-containing serum after different culture intervals (24, 48, and 72 h). The keratin hydrogel could obviously promote cell proliferation.
Biocompatibility and cell proliferation of HHK had been reported in some literatures. The hydrogels using keratins extracted from human hair by inducing polymerization with Ca2+ revealed that keratin hydrogels are comparable with collagen hydrogels in terms of the promotion of cell adhesion, proliferation, and the preservation of cell viability.16,30 Francesca investigated the surface adsorption of HHKs onto tissue culture polystyrene surfaces. These results suggest that HHKs can be used as a viable surface coating material to enhance substrate compliance for culturing cells. 31 According to our work, the results showed that the keratin hydrogel obviously promoted cell proliferation for only 72 h culture time. The cell proliferation for L929 murine fibroblasts also could be up to 10% under condition of weak gelation function of the keratin for only 24 h culture time. It can be predicted that the effect of the keratin hydrogel on cell will be more apparent under longer culture time.
Histology
The histological figures of 0.3 g keratin hydrogel implanted subcutaneously for 4 and 7 days are shown in Figure 7. For Figure 7(a) to (c) for 4 days, the epidermis and dermis were no obvious abnormalities. The keratins onto the subcutaneous muscle layer were yet not degraded completely (shown in white arrow) and the degradation site had a spotty lymphocytic infiltration (50–100/high power field (HPF)). The subcutaneous muscle layer had fiber medial necrosis (shown in red arrow) and the fibrous capsule had been formed around them. For Figure 7(d) to (f) for 7 days, the epidermis and dermis were also no obvious abnormalities and the keratin hydrogels could not be observed obviously. The subcutaneous muscle layer had fiber hyperplasia belt, including more capillary proliferation (4–7/HPE) (shown in black arrows) and a small amount of lymphocytic infiltration (⩽25/HPE).
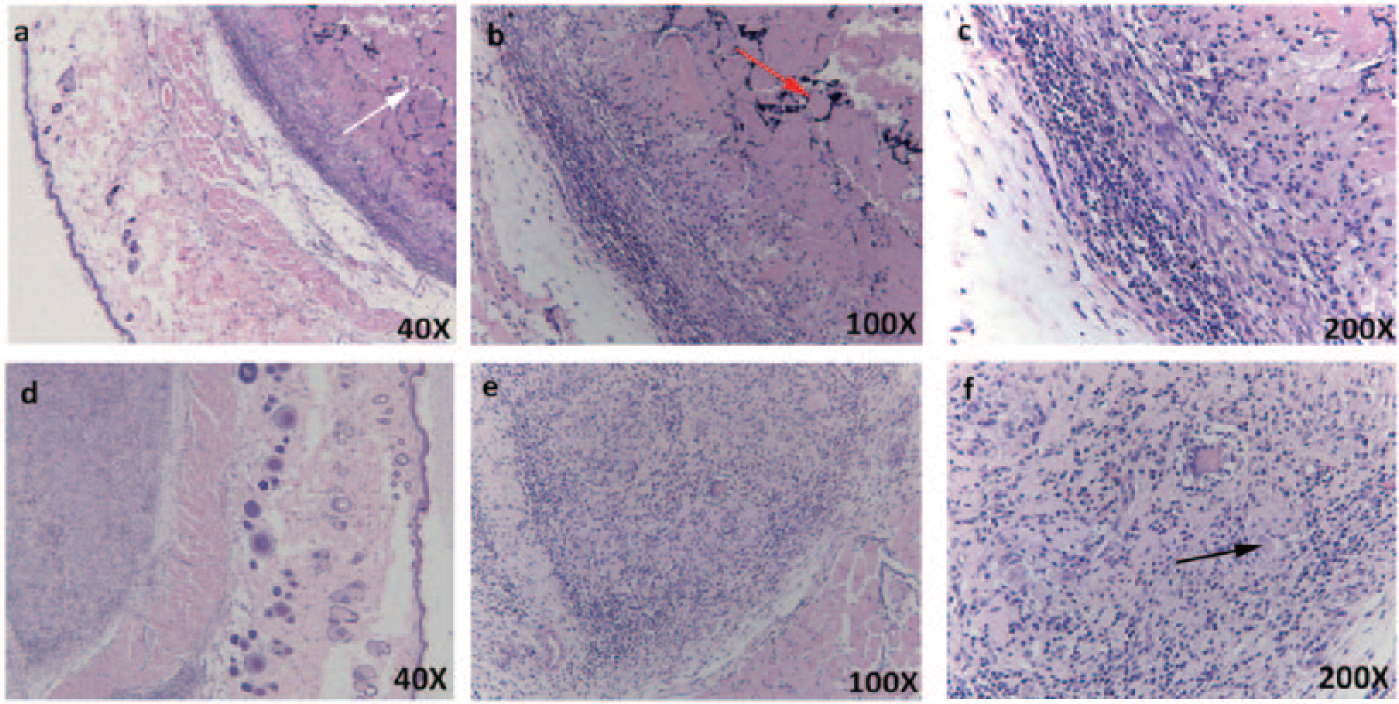
Histological sections of the keratin hydrogel subcutaneously implanted for 4 days ((a) to (c)) and 7 days ((d) to (f)). White arrow (a) indicates no degradation. Red arrow (b) indicates fiber medial necrosis. Black arrow (f) indicates more capillary proliferation.
The hydrolysis of the keratin hydrogel in vivo mainly depended on the function of enzyme and then the keratin hydrogel was absorbed.2,32 In our work, the keratins could be degraded in a week. Although the keratin degradation might cause inflammation and fiber necrosis, the tissue showed no obvious abnormalities and the keratin hydrogels still remained good absorption and promoted cell proliferation after a week.
Conclusion
In the work, the keratins were extracted from human hair using a modified method. In summary, the modified extraction method had two advantages as follows: (1) the modified reduction method was used to increase the molecular weight of the HHK and provide the self-assembly crosslinked ability of the keratin and (2) some crosslinked agents were not used to prepare the keratin hydrogels and some residues were avoided. In addition, the injectable keratin hydrogels prepared had high water absorption, good cell compatibility, and cell proliferation. The subcutaneous injection for SD rats confirmed that the keratin hydrogels had also good biocompatibility and could promote the formation of angiogenesis. As an allograft material, the HHK hydrogels are expected to be applied further in the field of skin wounds and some other soft tissue defect repair.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The authors are grateful for the financial support of the National Nature Science Fund of China (31000446) and Guangdong University Student Innovation Test Plan (1055914100).