
Abstract
One strategy for improving gene vector properties of polyethylenimine is to facilitate individual transfection mechanism steps. This study investigates (i) improving transfection efficiency by attaching peptide nuclear localization signals (nuclear localization signals: SV40 large T antigen nuclear localization signal or C-terminus of histone H1) to polyethylenimine (10 kDa) and (ii) using disulfide linkages, which are expected to be stable during polyplex formation, but cleaved inside cells giving improved gene release. Nuclear localization signal-containing polyplexes exhibited low cytotoxicity, whereas transfection efficiency with high molecular weight plasmid DNA increased up to 3.6 times that of underivatized polyethylenimine in Neuro2A cells at higher molar ratio of polyethylenimine-nitrogen to DNA-phosphate (N/P) ratios. However, with luciferase-specific low molecular weight small interfering RNA in Neuro2A/EGFPLuc cells, nuclear localization signal-containing polyplexes with disulfide linkages caused substantial cytotoxicity at N/P ratios >15 and no consistent significant reduction in luciferase expression. Possible explanations for molecular weight-dependent differences in genetic information transfer by polyplexes containing disulfide-linked nuclear localization signals are discussed.
Introduction
Before gene therapy can be widely used to address medical problems, some major drug delivery obstacles need to be surmounted. Viral vectors are the most efficient gene delivery agents, but they have serious problems in the areas of safety, immunogenicity and mutagenesis. 1 Non-viral vectors are substantially less efficient at gene delivery, but they have the advantages of being inherently safer and of being chemicals for which there are standard, well-accepted procedures for proving safety. 2 As a result, research in many laboratories has focused on ways to improve the transfection efficiency of non-viral vectors without increasing toxicity.
Gene delivery is a multistep process. Therefore, an effective vector must be multifunctional with different components designed to promote one or more of the various steps in the transfection process. 3 Among non-viral vectors, polymeric gene delivery carriers with modifiable groups in their backbones have proven to be useful scaffolds on which to add various structural elements that carry out the steps required for an effective non-viral gene delivery system.1,3,4 The most widely used polymeric gene carriers have employed commercially available materials such as polyethylenimine (PEI). PEI is relatively simple and inexpensive with a high amine content, which both facilitates chemical modification and accomplishes two or more steps in gene transfer processes.5–7 PEIs with varying molecular weights have been investigated, as have chemical modifications that vary the hydrophilic–hydrophobic balance of PEI in efforts to achieve higher transfection efficiencies without increasing cytotoxicity.7–10 Other modifications have included altering the surface charge density of PEI10,11 and adding targeting ligands.12–19
Some strategies for improving the effectiveness of non-viral vectors have focused on individual steps expected to be part of the gene delivery mechanism. The final steps involving translocation into the nucleus and insertion into host DNA have been particularly difficult to address, because little is known about the mechanisms of the natural processes involved. In the present study we have focused on improving gene delivery in the final steps by facilitating (i) translocation of a reporter gene to the nucleus and (ii) release of the gene from the vector prior to insertion into host DNA. A strategy that has been investigated for facilitating translocation of reporter genes to the nucleus is the addition of a nuclear localization signal (NLS) to either plasmid DNA or cationic polymers to facilitate passage through the nuclear envelope, believed to be a major limiting barrier in transfection of mammalian cells.20–22 NLSs are proteins and peptides that have the advantages of ease of production and use and high purity and homogeneity. These properties make them appropriate candidates for use in gene transfer systems alone or with other vectors. An example of a NLS is the basic peptide derived from the simian virus 40 large tumor antigen (Pro-Lys-Lys-Lys-Arg-Lys-Val) (SV40 NLS), which has been used to increase nuclear import of DNA, resulting in increased protein expression.23–25
Nuclear proteins such as histones are also known to have NLSs that facilitate their nuclear import. 26 Histones are believed to improve transfection by acting as DNA condensing agents. 22 It was found that the DNA condensation ability of histone H1 is higher than histones H2A, H2B, H3 and H4. 27 Histone H1 has been used to transfect DNA, particularly when used with calcium chloride and chloroquine. 28 Histone H1 has three domains, the N-terminal nose, the globular domain and the basic C-terminal tail. 29 The DNA condensation property of histone H1 has been localized to its C-terminal tail, 30 which has been shown to transfect DNA into a variety of mammalian cells with minimal toxicity. 31 Although short peptides are expected to have lower toxicity and immunogenicity than full length proteins, they are not always suitable candidates for use as a whole gene delivery system. In the present study, we have investigated the potential of NLSs for enhancing translocation of a reporter gene to the nucleus by preparing a series of branched 10 kDa PEI-based gene vectors with covalently bound small peptide nuclear localization signals with the sequences CCGPKKKRKV (SV40 large T antigen NLS; PEI–NLS) and CCGATPKKSTKKTPKKAKK (S/TPKK-containing octapeptide repeat motif present in the C-terminus of histone H1; PEI-H1). The same peptide sequences were also covalently attached to the hexanoate derivative of PEI, which was found in previous studies in this laboratory 10 to be the optimal alkyl derivative. The resulting conjugates were named PEI-6-8.5%-NLS and PEI-6-8.5%-H1, respectively.
One step in the PEI-mediated transfection mechanism that is particularly poorly understood is how plasmid DNA is released from its strong electrostatic interaction with PEI prior to insertion into host DNA. One explanation is that the DNA that gets inserted is tails or loops, from parts of the DNA–PEI complex where DNA is not directly interacting with PEI. Studies on the structures of DNA condensed on PEI and on other effective polycationic gene vectors indicate that much of the DNA is in the form of toroids that are not directly bound electrostatically to the polycation particle.32,33 In the present study we have investigated the potential for enhancing transfection efficiency of a reporter gene by facilitating its release from PEI prior to insertion into host DNA by attaching the nuclear localization signal through disulfide bonds. Disulfide bonds would be expected to be stable in the oxidizing environment present during formation of the nucleic acid–vector complex, but be cleaved in the reducing environment inside the cell. Added plasmid DNA is expected to associate with a NLS–PEI complex both through the NLS, which contains a preponderance of basic amino acids, and electrostatically with the PEI. The condensed DNA would be expected to keep the complex together even after the disulfide linkages have been cleaved, but the detached NLS should have greater flexibility to interact with cellular components, and this may facilitate gene translocation and insertion. As a negative control for high molecular weight DNA-stabilized polyplexes, we have selected polyplexes formed with small interfering RNA (siRNA). RNA interference utilizes 22-base-long double-stranded RNA segments, generated in nature by the enzyme dicer acting on high molecular weight double-stranded RNA. 34 The siRNA fragments would be expected to interact electrostatically with PEI along all or most of their 22-base length, leaving no functional tails available either for RNA interference activity or polyplex stabilization, as was the case with high molecular weight DNA. Double-stranded RNA electrostatically associated only with a NLS in the NLS–PEI complex may be released from the polyplex into the cytoplasm after endosomal escape and disulfide reduction, but siRNA electrostatically associated with PEI would not. Given the small size of the NLS, it is doubtful there would be enough NLS-associated siRNA to cause a statistically significant effect.
In the present study, we have prepared a series of vectors in which peptide NLSs are attached to 10 kDa PEI or to its hexanoate derivative through disulfide linkages. Branched 10 kDa PEI was selected as a scaffold for vector construction because of its low cytotoxicity, substantial transfection activity and conducive structural features, specifically large numbers of modifiable primary amine groups. The hexanoate derivative prepared by alkylating about 8.5% of the primary amine content of 10 kDa branched PEI with 6-bromohexanoic acid was also used as a scaffold, because it has been shown in this laboratory 10 to give increased transfection efficiency. The genetic information transfer activities of this series of disulfide-linked NLS-containing vectors have been compared with (i) high molecular weight DNA (which may have the capacity to stabilize polyplexes after reduction of disulfide linkages) and (ii) low molecular weight siRNA (which is expected to have less capacity to stabilize polyplexes, if any).
Materials and methods
Materials
The amino acid sequences CGGPKKKRKV (SV40 large T antigen NLS peptide) and CGGATPKKSTKKTPKKAKK (S/TPKK-containing octapeptide repeat motif present in the C-terminal of histone H1) were synthesized by CASLO Laboratory (Lyngby, Denmark). After synthesis, peptides were purified by high-performance liquid chromatography (HPLC; purity >95%) and sequences were confirmed by mass spectrometry. Branched PEI (bPEI; MW 10 kDa) was purchased from Polyscience, Inc (Warrington, PA, USA). The plasmid encoding luciferase under the control of cytomegalovirus (CMV) promoter (pRL-CMV-luc) and a luciferase assay kit were obtained from Promega (Madison, WI, USA). Ready to use siRNA duplexes were purchased from WGBiotech (Ebersberg, Germany), namely, GL3 luciferase-siRNA duplex; 5′-UUACGCUGAGUACUUCGAdTdT-3′; control-siRNA; nonspecific control duplex with similar GC content as anti-luciferase-siRNA; 5′-AUUGUAUGCGAUCGCAGACdTdT-3′. Dulbecco’s modified Eagle’s medium (DMEM) and fetal bovine serum (FBS) were purchased from GIBCO (Gaithersburg, MD, USA). Penicillin and streptomycin were purchased from Biochrom AG (Berlin, Germany). Ethidium bromide was purchased from Cinnagen (Tehran, Iran). Amicon Ultra® centrifugal membrane filter devices were obtained from Millipore (Billerica, MA, USA). Spectra/Por dialysis membranes were purchased from Spectrum Laboratories (Houston, TX, USA). All other solvents and reagents were purchased from Sigma-Aldrich (Munich, Germany).
Plasmid preparation
pRL-CMV-luc was propagated in chemically high-copy competent DH5-α Escherichia coli strain and extracted from overnight bacterial cultures with a Qiagen Endofree Mega Plasmid Kit (QIAGEN, Hilden, Germany) using standard Qiagen protocols. The purity of plasmid preparations was determined by the ratio of absorbance at 260 nm to that of 280 nm (A260/A280 ratios ≥1.8).
Synthesis of non-viral carriers
Preparation of 6-bromohexanoic acid derivative of PEI
Alkylation of PEI10 with 6-bromohexanoic acid was carried out as reported previously. 10 Briefly, 6-bromohexanoic acid (0.09 g, an amount calculated to alkylate 10% of primary amines) in 5 ml dimethylformamide (DMF) was added drop wise with stirring to a solution of 0.5 g PEI10 in 5 ml of DMF. The reaction mixture was stirred 48 h at room temperature, dialyzed in 8000 Da cut-off Spectra/Por dialysis tubing four times against water to remove unreacted alkylating agents, and lyophilized. The primary amine content of PEI preparations was determined by assay with trinitrobenzensulfonic acid (TNBS). 35
Modification of PEI10 and its 6-bromohexanoic acid derivative with 3-(2-pyridyldithio) propionic acid N-hydroxy-succinimide ester (SPDP)
PEI10 (5.3 mg) or the hexanoate derivative of PEI10 (6.3 mg) were treated with 1 mg of SPDP in 2 ml buffer (20 mM HEPES, 350 mM NaCl, pH 8) 36 for 2 h at room temperature under argon. Unreacted SPDP was removed using centrifugal membrane filter devices with 5 kDa molecular weight cutoff membranes that were conditioned before use with a solution of PEI10. The preparations were diluted with buffer and centrifuged in the filter devices for 30 min at room temperature, and the retentate diluted with buffer (20 mM HEPES, 250 mM NaCl, pH 7.4) and the process repeated twice. Aliquots of each preparation were flash frozen and stored at −80℃ until used. The degree of modification with SPDP was determined in an aliquot of each preparation by releasing 2-thiopyridone (DTP) by reduction for 30 min at room temperature with excess dithiothreitol (DTT). The DTP released was determined spectrophotometrically (extinction coefficient at 343 nm: 8.08 × 103 M−1 cm−1). The primary amine contents of PEI-DTP and hexanoate PEI-DTP were determined using TNBS. 35
Conjugation of peptides to thiol-functionalized PEI10
Peptides CGGPKKKRKV or CGGATPKKSTKKTPKKAKK (1 mg each in 200 µl of degassed deionized water) were added to aliquots of PEI-DTP in amounts calculated to achieve coupling of 10% of primary amines with peptides. The reaction mixtures were diluted to 2 ml with buffer (20 mM HEPES, 500 mM NaCl, pH 7.1) 36 and incubated 4 h at room temperature under argon. The amount of DTP released by reaction with thiol groups in the peptides was determined as described above and used to estimate the amount of peptide conjugated. Unreacted peptides were removed using five centrifugation runs of 30 min each at 4℃ in membrane filter devices with 10 kDa molecular weight cutoff membranes followed by redisolution of the retentate in buffer (20 mM HEPES, 50 mM NaCl, pH 7.4). Solutions of the peptide conjugates in the retentates were flash frozen in aliquots and stored at –80℃ until used.
Conjugates were characterized in aliquots in two ways. The unreacted amine content was determined with the TNBS assay. The presence of bound peptide was confirmed using RP-HPLC after peptide release from the conjugate by exchange with the free thiol groups in 2-mercaptoethanol (ME) during 30 min incubation at room temperature. Chromatograms obtained from each peptide-modified PEI before and after ME treatment were compared and the presence of a peptide peak after incubation with ME used to confirm coupling of peptides.
Polyplex preparation
Polyplexes were prepared in a range of N/P ratios (molar ratio of PEI-nitrogen to DNA-phosphate) by adding a range of concentrations of PEI or PEI-peptide conjugates in buffer with equal volumes of the same buffer containing RL-CMV-luc plasmid, mixing by gentle pipetting and incubating 30 min at room temperature.
Polyplex size
The average particle size of vector/pDNA complexes was determined using Dynamic Light Scattering (DLS) on a Malvern Nano ZS instrument (Malvern Instruments, UK). Scattered light was detected at 173° angle. The N/P ratio of polyplex preparations was varied by preparing a range of concentrations of vector in HBG buffer (HEPES-buffered glucose containing 20 mM HEPES, 5% glucose, pH 7.4), which were each added to an equal volume of DNA solution containing 5 µg plasmid in the same buffer, mixed by gently pipetting and incubated for 30 min at room temperature. Polyplex preparations were then diluted to a final volume of 1 ml in triplicate and light scattering measured.
Zeta potential
Zeta potential measurements of polyplexes were carried out using Laser Doppler Velocimetry (LDV) on a Malvern Nano ZS instrument. Vector/pDNA complexes were prepared as described for DLS measurements.
DNA condensation
DNA condensation in polyplexes by PEI or its derivatives was determined by ethidium bromide (EtBr) exclusion assay. EtBr access to DNA was determined by fluorescence (excitation λex: 510 nm and emission λem: 590 nm) in a Jasco FP-6200 fluorometer (Tokyo, Japan). Plasmid DNA (5 µg) was complexed with EtBr (400 ng/ml) in 1 ml HBG buffer and the fluorescence intensity measurement taken as the 100% value. The fluorescence intensity of 400 ng/ml EtBr solution was used as the background value. A range of concentrations of PEI or other derivatives (in 2.5 N/P ratio increments) was added to the fluorometer cuvette containing DNA and EtBr, followed by gentle mixing. Plasmid condensation by PEI or its derivatives was measured in triplicate as decreased fluorescence intensity and reported as mean ± SD.
Cytotoxicity
Neuro2A murine neuroblastoma cells (ATCC CCL-131, American Type Culture Collection, Manassas, VA, USA) were cultured at 37℃ in a humidified 5% CO2 atmosphere in DMEM supplemented with 10% FBS, 100 U/ml penicillin and 100 µg/ml streptomycin. The medium was replaced every 48 to 72 h, and cells were sub-cultured when they reached 60–80% confluency. The cytotoxicity of PEI and PEI-peptide conjugates were determined in triplicate using the MTT colorimetric assay. 37 Neuro2A cells were seeded in 96-well plates at an initial density of 1 × 104 cells per well in 100 µl DMEM medium with 10% FBS, cultured 24 h, cells and treated for 3 h with 25 µl of polyplex preparations in serum-free DMEM medium containing 200 ng pDNA at the same N/P ratios used for the transfection experiments. The medium was replaced with fresh DMEM medium containing 10% FBS and the cells cultured 18 h, followed by addition of 10 µl MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide, 5 mg/ml in phosphate buffered saline] solution per well and incubated for 3 h at 37℃. The medium was removed and the formazan formed was dissolved in 100 µl DMSO. Absorbance was measured at 545 nm on a STAT FAX-2100 microplate reader (Awareness Technology, Palm City, FL, USA) and cell viability estimated as percent absorbance of PEI-treated cells relative to untreated control cells.
Erythrocyte leakage
Erythrocytes were washed four times with phosphate-buffered saline (PBS) by centrifugation and resuspension of the pellet in 150 mM NaCl. Serial dilutions of PEI-peptide conjugates containing 10–80 µg/ml PEI in HEPES-buffered saline (HBS, 20 mM HEPES, 150 mM NaCl, pH 7.4) were prepared in triplicate and 10 µl of erythrocyte suspension added to each well in V-bottomed 96-well plates. The plates were incubated at 37℃ for 30 min with constant shaking, centrifuged (2000 rpm, 10 min) and 70 µl of the supernatant transferred to a flat-bottom 96-well plate. The amounts of hemoglobin released were measured at 405 nm using a microplate reader. A solution of 1% Triton X-100 was used as positive control representing 100% leakage.
Transfection efficiency
Neuro2A cells were cultured at a density of 1 × 104 cells per well in DMEM medium containing 10% FBS using 96-well plates for 24 h prior to transfection. Polyplexes were prepared with PEI or PEI-peptide conjugates in a range of N/P ratios in 250 µl DMEM. For transfection, 25 µl of the polyplex mixture was added to wells with Neuro2A cells in triplicate and incubated for 3 h at 37℃. The transfection medium was then removed, and the cells were cultured under the same conditions in a complete medium for 18 h. The medium was removed and cells in each well were lysed with 50 µl Promega lysis buffer. Luciferase activity in 20 µl of cell lysate was quantified by mixing with 100 µl of luciferase assay reagent from a Luciferase assay kit (Promega, Madison, WI, USA), and luminescence measured on a Sirius Tube Luminometer (Berthold Detection Systems, Pforzheim, Germany). Measurements were reported as relative luminescence units (RLU) per number of seeded cells.
siRNA transfection
Neuro2A/EGFPLuc cell line (a Neuro2A mouse neuroblastoma cell line mouse neuroblastoma stably expressing luciferase) was kindly provided by Prof. E. Wagner (Department of Pharmacy, Ludwig Maximilians University, Munich, Germany) and used for siRNA transfection studies. siRNA transfection with PEI or PEI-peptide conjugates was determined in triplicate in 5000 Neuro2A/EGFPLuc cells per well of a 96-well plate cultured for 24 h in DMEM with 10% FBS, 100 U/ml of penicillin and 100 µg/ml of streptomycin and maintained at 37(℃ and 5% CO2. Immediately before transfection, the culture medium was removed from the wells and replaced with 80 μl fresh growth medium containing 10% FBS. Transfection complexes containing 500 ng of luciferase-specific siRNA (LucsiRNA) in HBG in a range of N/P ratios were added to each well and incubated at 37(℃ without medium change. After 48 h, luciferase activity was measured in a luminometer (Lumat LB9507 instrument, Berthold, Bad Wildbad, Germany) in 20 μl of cell lysate mixed with 100 μl luciferase assay buffer from a luciferase assay kit (Promega, Mannheim, Germany). Control transfections were carried out with a non-specific siRNA (MutsiRNA) in order to distinguish between specific gene silencing and unspecific knockdown of protein expression due to carrier cytotoxicity.
Statistics
Experiments were conducted in triplicate or quadruplicate and the data analyzed by comparing experimental results to the appropriate controls using Student’s t-test. P values ≤0.05 were considered statistically significant.
Results
Synthesis of non-viral vectors
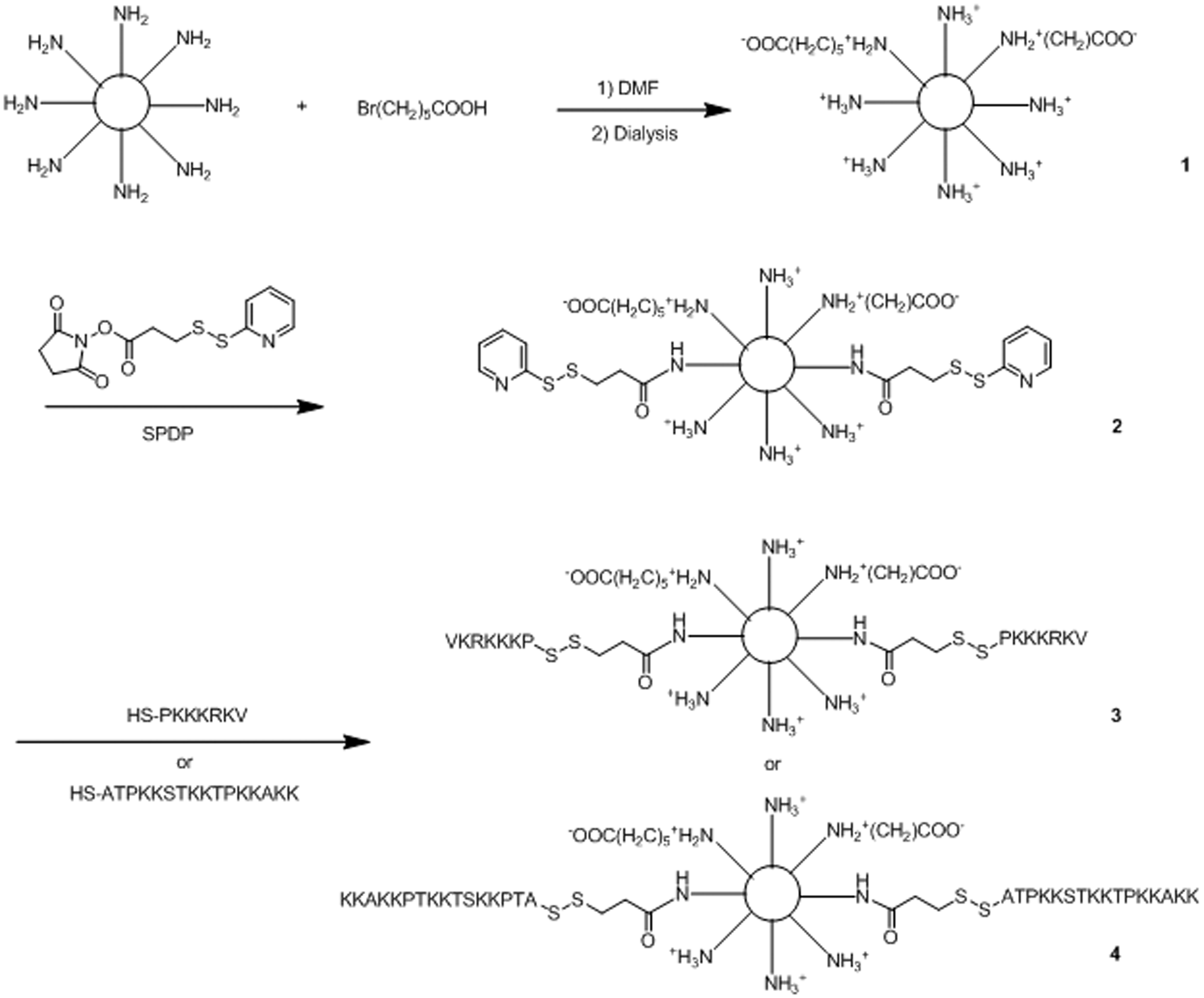
Branched PEI (10 kDa) was used as a scaffold on which to synthesize vectors (Schemes 1 and 2). PEI was initially alkylated with 6-bromohexanoic acid (six-carbon alkyl chain) on 8.5% of primary amines (based on TNBS assay values). Solutions of PEI or the 6-bromohexanoic acid derivative were activated with SPDP, and then derivatized with peptides added at amounts calculated to give 10% primary amine conjugation. Subsequent steps of the synthesis were carried out on freshly derivatized PEI. It was estimated that about 9% of primary amines were modified with peptides based on the release of free 2-thiopyridone during peptide coupling. The attachment of peptides was confirmed by treating a sample of the preparation with ME to release free peptide, which was then detected by HPLC (Supplementary Figure). Vector purification was carried out by centrifugation in membrane filter devices followed by redissolution of the retentate, which was repeated twice. The resulting vectors were identified as PEI–X–Y%–Z, in which X is the number of carbons in the alkyl chain, Y% is the percentage of primary amines substituted with alkyl chains and Z represents the peptide conjugated to the modified PEI structure. As a result, the following products were obtained: (a) PEI-NLS, (b) PEI-alkyl-NLS, (c) PEI-H1 and (d) PEI-alkyl-H1 (Schemes 1 and 2).
The chemical synthetic methods used to conjugate PEI10 to NLS and H1 peptides. (1) PEI-PDP (activated PEI), (2) PEI-NLS and (3) PEI-H1. NLS: nuclear localization signal; PEI: polyethylenimine. The chemical synthetic process used to make PEI-alkyl-peptide. (1) PEI-alkyl, (2) PEI-alkyl-PDP, (3) PEI-alkyl-NLS and (4) PEI-alkyl-H1. NLS: nuclear localization signal; PEI: polyethylenimine.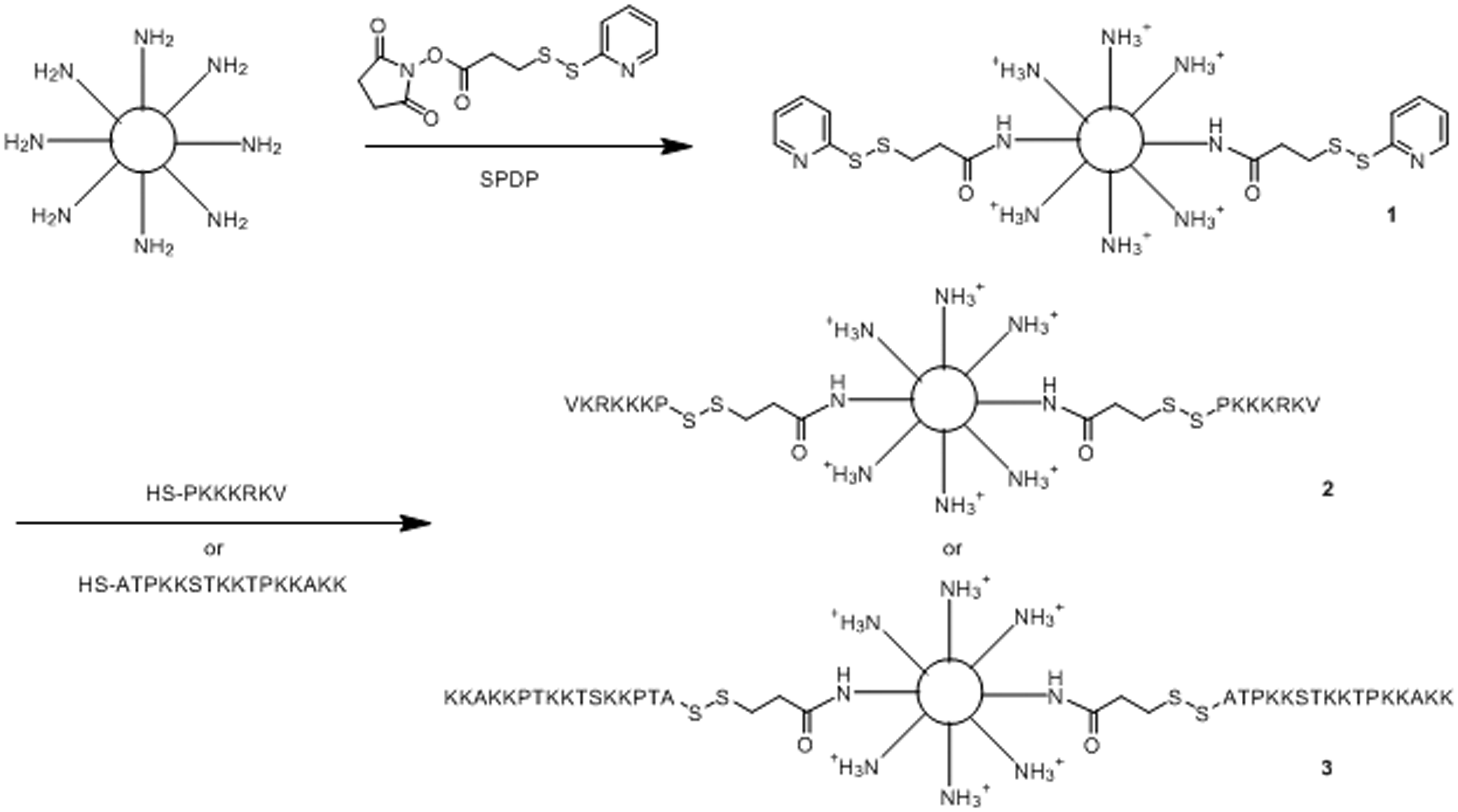
Polyplex size
Polyplex size has been observed
38
to be a very important factor affecting cellular uptake for some cell lines in which only submicron sized particles were efficiently taken up. Particles in the submicron size range may have uptake mechanisms like caveolae-mediated endocytosis in addition to the classical clathrin-mediated endocytosis.9,39 Dynamic light scattering was used to measure the size of polyplexes prepared across the range of N/P ratios used in this study. Unmodified PEI10, which was used as a control yielded polyplexes with diameters of 45.1 ± 5.3 nm (Figure 1). As expected, adding alkyl chains resulted in polyplexes with a larger mean size of about 71.2 ± 6.1 nm for PEI-6-8.5%, 76.8 ± 5.2 nm for PEI-6-8.5%-H1 and 41.9 ± 6.8 nm for PEI-6-8.5%-NLS, whereas conjugating PEI with peptides to get PEI-H1 and PEI-NLS resulted in polyplexes with decreased particle size of about 60 nm for PEI-H1 and 27 nm for PEI-NLS. Thus, the vectors prepared in this study can condense pDNA into particles in the submicron size range optimal
5
for gene delivery.
Average particle sizes and zeta potentials of different polyplexes at N/P ratio of 15. *(P ≤ 0.05), size of hexanoated PEI-peptide compared to PEI-peptide. N/P ratio: molar ratio of PEI-nitrogen to DNA-phosphate; PEI: polyethylenimine.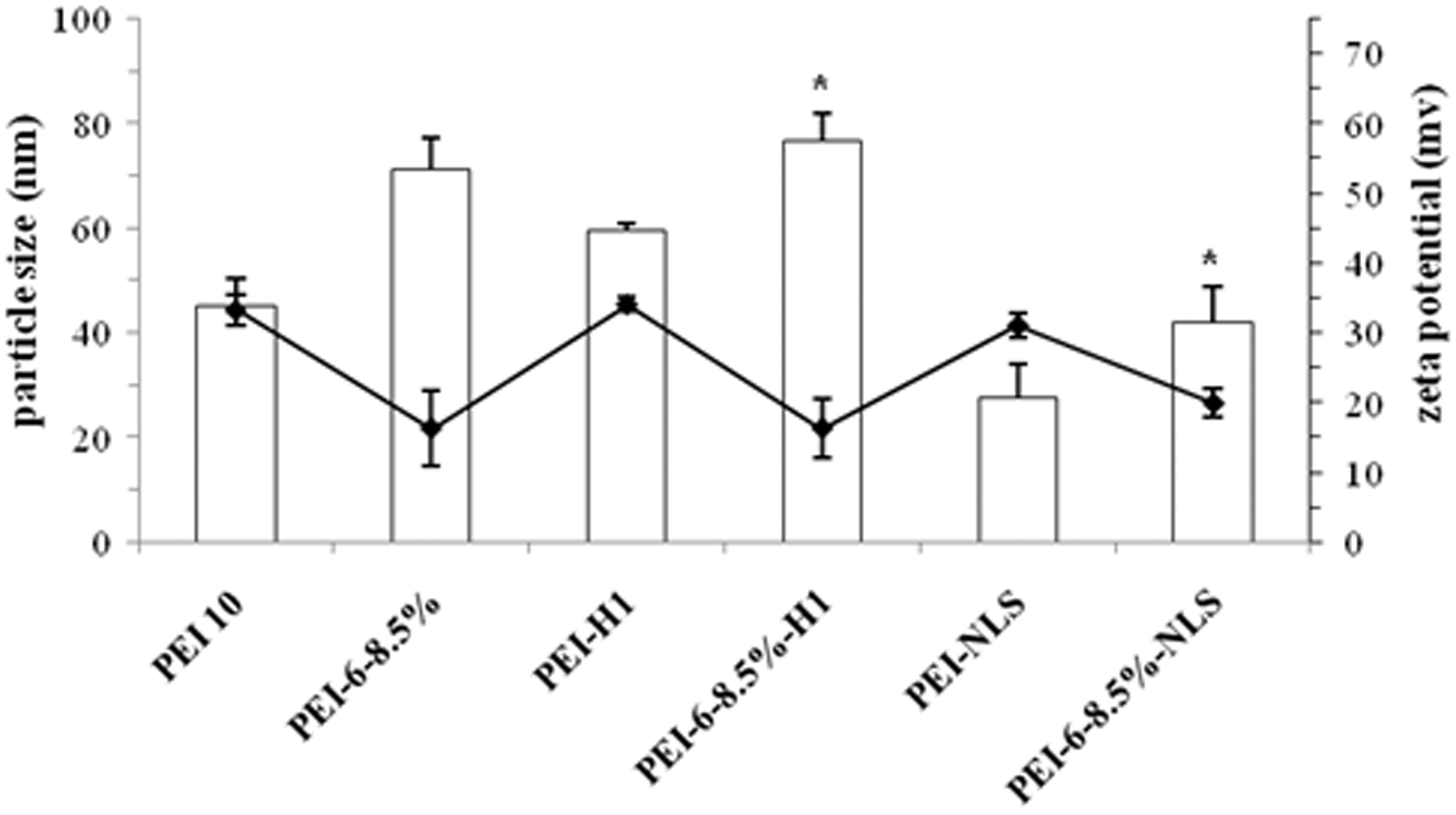
The ζ-potentials of the vector/DNA complexes were in the range 15–35 mV (Figure 1), indicating that all nanoparticles had sufficient positive charge after DNA complexation to both prevent aggregation through electrostatic repulsion and to bind to the negatively charged cell surface. 9 Although the peptides used in this study had considerable arginine content, coupling them to either PEI or its hexanoate derivatives did not significantly alter the surface positive charge, although the series of hexanoate derivatives all had significantly reduced surface charge compared to the corresponding PEI derivative, similar to previous observations with alkanoate chain grafting to PEI.5,10
DNA condensing ability
Vectors must condense DNA into nanoparticles for efficient delivery into cells. This condensation results from electrostatic attractions between pDNA and the polycationic vector. Tightly bound DNA cannot expand enough to allow intercalation by ethidium bromide (EtBr), a dye that fluoresces only when intercalated. Thus, formation of tightly condensed DNA in polyplexes by modified and unmodified PEI can be observed as reduced fluorescence intensity in the presence of excess EtBr. Results from the EtBr exclusion assay (Figure 2) showed that all vectors prepared in this study were able to efficiently bind plasmid DNA. For PEI10, optimal DNA condensation (EtBr fluorescence intensity reduced by more than 80%) occurred at an N/P ratio of 2.5 with no significant increase in DNA condensation at higher N/P ratios. For PEI10 derivatized with alkylcarboxylate and/or a peptide, higher N/P ratios were needed to achieve the same optimal DNA condensation level. However, at N/P ratios ≥15, all vectors exhibited optimal DNA condensation.
DNA condensation ability of polycationic vectors measured by EtBr assay. EtBr: ethidium bromide.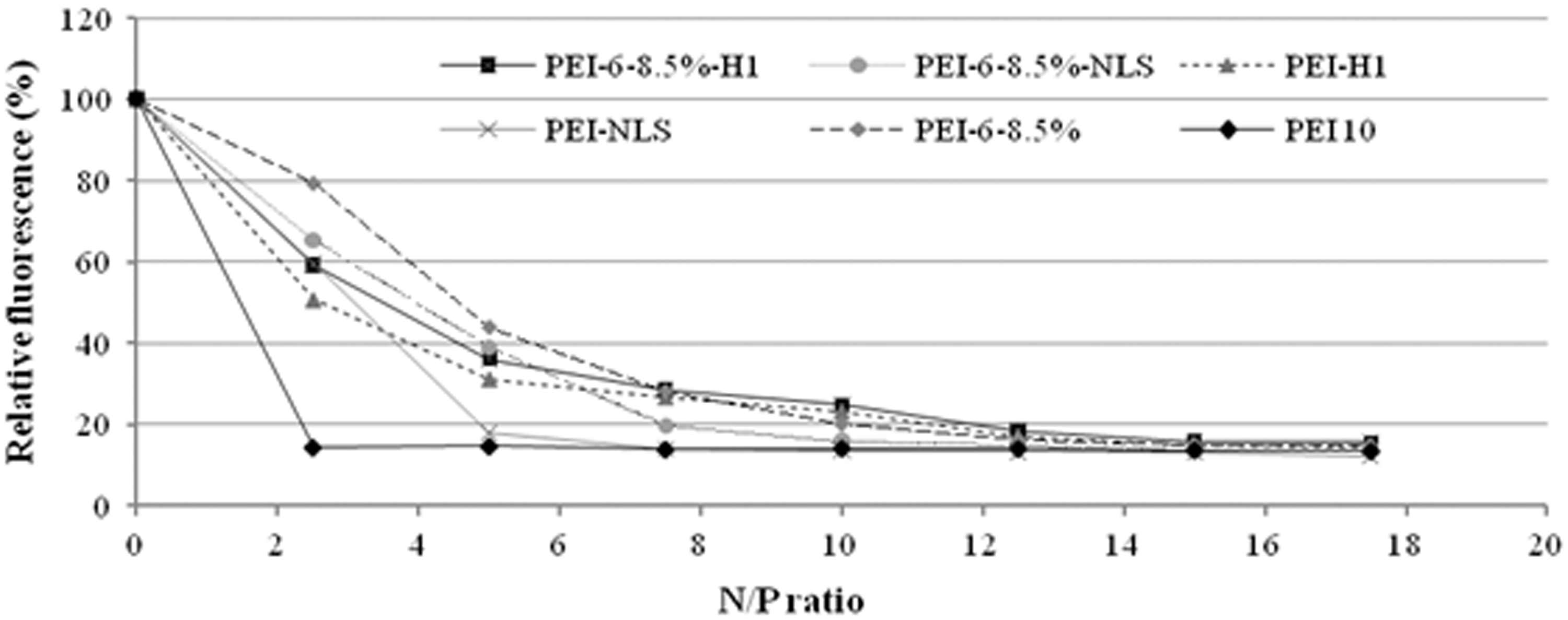
Cytotoxicity
Hemolytic activity was used to assess the membrane destabilizing properties of PEI and the derivatives prepared from it prior to complexation with DNA. The percent erythrocyte lysis by PEI was less than 15% at pH = 7.4 and the hemolytic activity of peptide hexanoate derivatives of PEI was generally less than that of PEI itself (data not shown). Reduced hemolytic activity following hydrophobic modification of PEI has been reported previously. 40
Cytotoxicity of polyplexes prepared from plasmid DNA and PEI or its derivatives was also examined in cultures of Neuro2A cells. There was no significant cytotoxicity observed for polyplexes prepared from PEI derivatives containing attached H1 at any N/P ratio tested (Figure 3). Attaching NLS to PEI or the hexanoate derivative was less effective at reducing cytotoxicity of the resulting polyplexes. All vectors used in this study resulted in polyplexes with relatively low cytotoxicity (viability typically >90%) when complexed with plasmid DNA at an N/P ratio of 5, which is an effective ratio for use in transfection studies (see below). Higher N/P ratios resulted in polyplexes with greater cytotoxicity, but no preparation resulted in viability less than 75%.
Viability of Neuro2A cells exposed for 3 h to polyplexes prepared from plasmid DNA and PEI10 or modified vectors prepared from it at the indicated N/P ratios. Cell viability was measured 18 h later as formazan color formation from MTT relative to untreated control cultures, which were defined as 100% viable. Statistically significant differences in cell viability between polyplexes prepared with PEI10 and a modified PEI10 are indicated as *(P ≤ 0.05). N/P: molar ratio of PEI-nitrogen to DNA-phosphate; PEI10: polyethylenimine.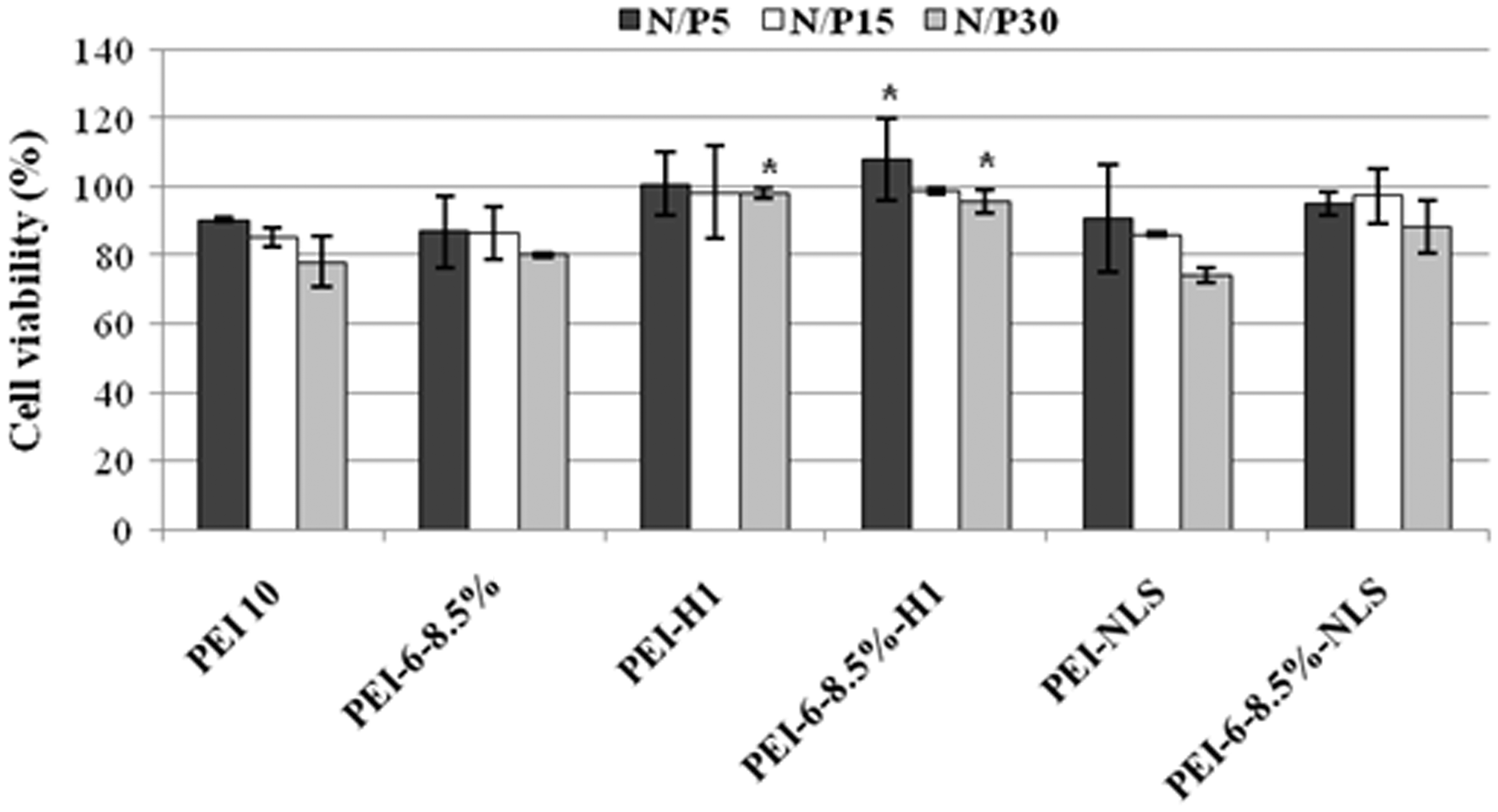
Transfection activity
Transfection activity of PEI and the derivatives prepared from it was evaluated in Neuro2A cells using polyplexes prepared with plasmid DNA coding for Renilla reniformis luciferase. Expression of luciferase enzyme activity was measured using a commercial assay kit. Transfection of pDNA in the absence of a polycation vector resulted in undetectable luciferase expression (data not shown). All derivatives of PEI tested resulted in significantly higher transfection efficiencies than unmodified PEI for at least two N/P ratios (Figure 4). The most effective transfection was observed with histone H1 coupled to PEI, which at a N/P ratio of 30 achieved an about 3.6-fold increase compared to PEI10 at the same N/P ratio (Figure 4).
Transfection efficiency of polyplexes prepared from plasmid DNA encoding the luciferase gene from sea pansy (Renilla reniformis) and PEI and the derivatives prepared from it. Luciferase activity expressed in Neuro2A cells after 18 h following a 3-h treatment with polyplexes was measured in cell lysates as light emitted in relative luminosity units (RLU). Statistically significant differences in transfection efficiency of modified PEI compared to PEI10 are indicated as *(P ≤ 0.05). PEI: polyethylenimine.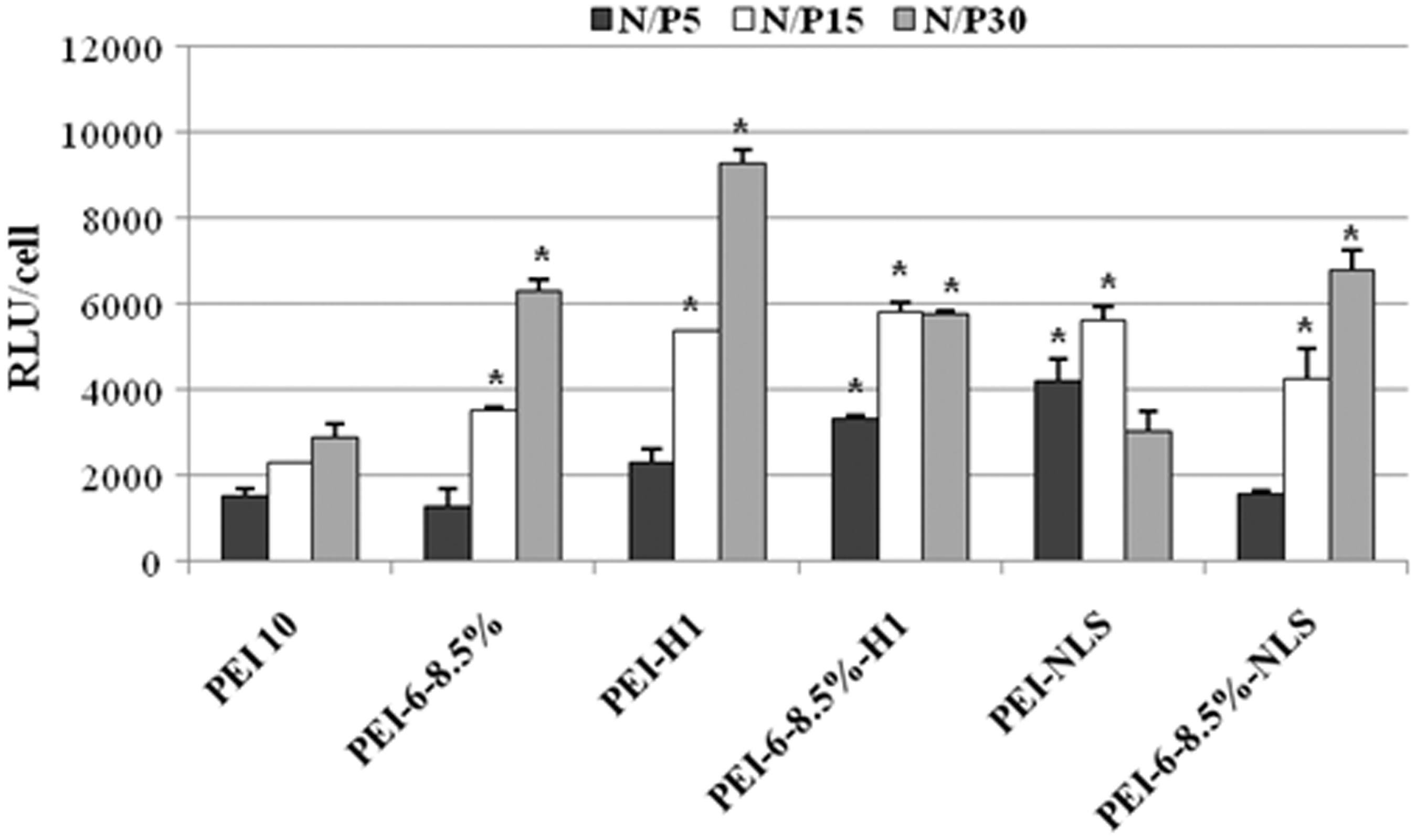
PEI grafted with SV40 large T antigen NLS also formed polyplexes with significantly enhanced gene expression levels relative to polyplexes formed with unmodified PEI, but the increase in transfection activity was less than obtained with PEI grafted to H1 (Figure 4).
As observed previously, 10 altering the hydrophilic–hydrophobic balance of PEI by forming hexanoate derivatives resulted in polyplexes with significantly higher transfection efficiency than unmodified PEI at the higher N/P ratios studied (Figure 4). However, coupling nuclear localization peptides to the hexanoate derivative of PEI did not significantly increase the transfection efficiency of polyplexes relative to the unmodified hexanoate derivative of PEI in the case of either H1 or the SV40 large T antigen NLS.
As shown in Figure 5, there was no significant reduction in luciferase expression with luciferase-specific siRNA and any of the vectors studied at the lowest test concentration, indicating no detectable toxic effects of the vectors on luciferase expression at the lowest N/P ratio. For both unmodified PEI and hexanoate-modified PEI, luciferase expression was reduced at higher N/P ratios, but the same degree of reduction occurred with LucsiRNA as with MutsiRNA, indicating that the reductions were the result of toxicity. Coupling nuclear localization signals, particularly H1, resulted in less toxicity at higher N/P ratios, but there was no consistent increased reduction in luciferase expression associated with LucsiRNA relative to MutsiRNA. Significantly reduced luciferase expression would have been expected for siRNA interference by the siRNA associated with the increased H1 or NLS.
Transfection activity of PEI-containing polyplexes prepared with luciferase-specific siRNA (LucsiRNA, light-colored bars) in Neuro2A/EGFPLuc, a Neuro2A mouse neuroblastoma cell sub-line stably expressing luciferase. As a control for cytotoxicity caused by PEI vectors, parallel cultures of Neuro2A/EGFPLuc cells were transfected with polyplexes prepared in the same manner with nonspecific siRNA (MutsiRNA, dark-colored bars). Luciferase activity in 20 μl of cell lysates was measured with a luminometer. Luciferase activity in parallel cultures not treated with polyplexes was measured as the 100% control. PEI: polyethylenimine; siRNA: small interfering RNA.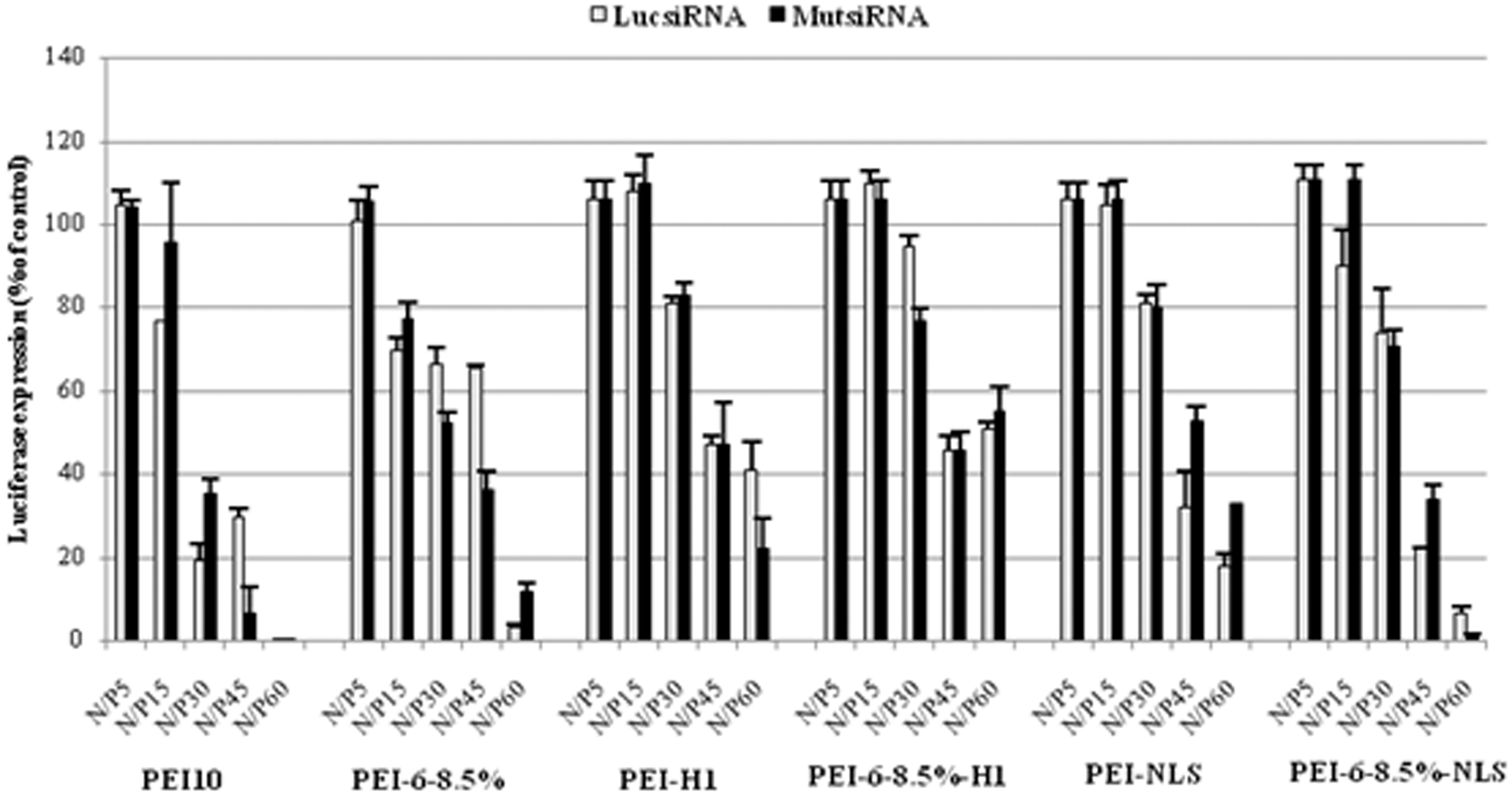
Discussion
In the present study, to investigate the potential of nuclear localization signals for enhancing the transfection efficiency of a reporter gene, we synthesized a series of PEI-based gene delivery vectors modified with NLSs. Small peptide nuclear localization signals with the sequences CCGPKKKRKV and CCGATPKKSTKKTPKKAKK were bound through disulfide bonds to either PEI (resulting in conjugates named PEI-NLS and PEI-H1, respectively) or to PEI-hexanoate (resulting in conjugates named PEI-6-8.5%-NLS and PEI-6-8.5%-H1, respectively).
Nuclear localization sequences, covalently or noncovalently conjugated to DNA or to DNA complexing polymers, have been reported to increase the DNA delivery into the nucleus23,25,41 using the nuclear import machinery. 42 NLS-containing peptides incorporated into polyplexes may also interact with proteoglycans to enhance cell entry of the heteroplex and possibly interfere with other steps of the gene transfer mechanism. 21 It has been reported that extended domains of basic amino acid residues in both core and H1 histones mediate nuclear transport by mechanisms similar to, but not identical with the classical pathway, whereas nuclear transport by globular histone appeared to require interactions with transport receptors based on conformational information rather than short sequences. 26 The enhanced transfection efficiency observed in this study with the vector PEI-H1 is consistent with the H1 C-terminus acting as a NLS by a mechanism described by Baake et al. 26 However, another study claimed that fragments of the C-terminal domain of H1 that were smaller than 20 amino acids could not function as an NLS. 43
A possible reason for higher transfection activity of H1-modified PEI could be the greater DNA condensing effect of this peptide,29,30 which would hold the polyplex together after reduction of disulfide linkages, or more efficient nuclear localization, or a combination of the two effects.
Although alkylation of PEI resulted in polyplexes with higher transfection efficiencies at the higher N/P ratios, but it is unclear why the enhanced transfection efficiency of polyplexes observed with carboxyalkylation is not at least additive with the enhanced transfection efficiency of polyplexes obtained by conjugation of nuclear localization peptides, because they are believed to act by different mechanisms.
Carlisle et al. 44 using adenovirus hexon protein as the nuclear localization component observed that transfection levels in HepG2 cells were more than twice as great with the NLS coupled to PEI through a disulfide linkage than through a thioether linkage. They offered the following two possible explanations for how enhanced transfection could occur despite attachment of the nuclear localization signal to the DNA through a linkage expected to be cleaved inside cells: (i) nuclear translocation could occur before disulfide reduction did and (ii) the reducing potential is stronger in the nucleus than the cytoplasm 45 so that disulfide linkages are stable in the cytoplasm but cleaved in the nucleus. The rationale for the present study is that high molecular weight plasmid DNA may hold polyplexes together after disulfide cleavage in a form allowing more effective interaction with host cell nuclear translocation and/or DNA-release mechanisms, offers an alternative explanation for the observations of Carlisle et al. 44
Transfection activity with siRNA associated with the same PEI-peptide scaffolds was examined as a strategy for introducing siRNA, or at least the part of it associated with the cationic histone or with the NLS, into the cytoplasm, after the PEI component had mediated endosomal escape and the disulfide linkages had been cleaved by the reducing environment inside the cell. Use of complexes with siRNA also serves as a negative control for transfection with a nucleic acid large enough to hold polyplexes together in the absence of disulfide linkages, because the 22-nucleotide-long siRNA pieces would not be expected to be large enough to hold the complexes together after disulfide reduction.
PEI has been reported by several laboratories to be effective at siRNA delivery,46,47 whereas other laboratories have observed that PEIs with high efficiency in plasmid DNA transfer were not effective at siRNA delivery.48,49 In one study, some factors such as the relatively weak binding affinity and biophysical and structural characteristics of a given PEI structure were stated as possible reasons for poor siRNA transfection of these conjugates. 49 It has also been suggested that some PEI structures form complexes with siRNA that are so stable that free siRNA cannot be released in the cytoplasm. 48 It would be necessary to have competition between PEI and natural polyamines like spermine for interaction with the exogenous nucleic acids in order to release it from the conjugates. It is believed that release by this mechanism would not occur at an efficient rate in the cytoplasm, where siRNA exerts its biological effect. 48 The lack of observable RNA interference in the current study may be a result of siRNA associated with PEI not being released in the cytoplasm and an insufficient amount of siRNA associated with H1 or SV40 NLS peptides to cause an observable effect, even if released. Alternatively, siRNA released with the peptides may have become electrostatically bound to the PEI and thus unavailable.
Conclusion
We synthesized a series of PEI derivatives in which a NLS (SV40 large T antigen NLS or the S/TPKK-containing octapeptide repeat motif present in the C-terminus of histone H1) was attached to PEI10 or PEI10-hexanoate through a disulfide linkage. For transfection of plasmid DNA encoding luciferase, disulfide-linked SV40 NLS significantly enhanced transfection efficiency when attached to PEI10, but not when attached to PEI10-hexanoate. Disulfide-linked histone H1 NLS significantly enhanced DNA transfection for both PEI10 and PEI10-hexanoate at lower N/P ratios. These results confirm reports from other laboratories that including NLSs in gene delivery systems enhances DNA transfection and that high levels of transfection are obtained using a disulfide linkage. In contrast, transfection of luciferase-specific siRNA did not significantly reduce luciferase expression with any of the vectors studied. One explanation for the effectiveness of this system in the transfection of relatively long DNA sequences but not short dsRNA sequences is that high molecular weight DNA can stabilize polyplexes containing disulfide-coupled NLSs in a manner that allows enhanced transfection efficiency. These vectors may have a potential for in vivo use.
Footnotes
Acknowledgements
The siRNA experiments were performed at Pharmaceutical Biotechnology Center of Drug Research, Ludwig-Maximilians-University, Munich, Germany, under supervision of Alex Philipp and Prof. Ernst Wagner.
Funding
This work was supported by Mashhad University of Medical Sciences, Mashhad, Iran and the Iranian Nanotechnology Initiative.