
Abstract
A composite of 70/30 poly(lactic acid)/hydroxyapatite was systematically prepared using various amounts of glycidyl methacrylate as reactive compatibilizer or Joncryl ADR®-4368 containing nine glycidyl methacrylate functions as a chain extension/branching agent to improve the mechanical and biological properties for suitable usage as internal bone fixation devices. The effect of glycidyl methacrylate/Joncryl on mechanical properties of poly(lactic acid)/hydroxyapatite was investigated through flexural strength. Cell proliferation and differentiation of osteoblast-like MC3T3-E1 cells cultured on the composite samples were determined by Alamar Blue assay and alkaline phosphatase expression, respectively. Result shows that flexural strength tends to decrease, as glycidyl methacrylate content increases except for 1 wt.% glycidyl methacrylate. With an addition of dicumyl peroxide, the flexural strength shows an improvement than that of without dicumyl peroxide probably due to the chemical bonding of the hydroxyapatite and poly(lactic acid) as revealed by FTIR and NMR, whereas the composite with 5 wt.% Joncryl shows the best result, as the flexural strength increases getting close to pure poly(lactic acid). The significant morphology change could be seen in composite with Joncryl where the uniform agglomeration of hydroxyapatite particles oriented in poly(lactic acid) matrix. Addition of the epoxy functional compatibilizers at suitable percentages could also have benefits to cellular attachment, proliferation, differentiation and mineralization. So that, this poly(lactic acid)/hydroxyapatite composite could be a promising material to be used as internal bone fixation devices such as screws, pins and plates.
Keywords
Introduction
Biomaterials, for example, hydroxyapatite, bioglass, polymers and metal, have been used as a bone substitution during the past 40 years. Particularly, an internal bone fixation device being used clinically in metallic devices which are now replaced by biodegradable polymer/bioceramic composite such as poly(lactic acid)/hydroxyapatite due to needless post-operation after bone healing. Among biodegradable polymers, poly(lactic acid) (PLA) is of great interest to researchers due to its excellent biodegradability and mechanical properties, therefore it is widely use in biomedical applications particularly for bone fixation material.1–4 Hydroxyapatite (HA) (Ca10(PO4)6(OH)2) is a calcium phosphate (CaP)-based compound which is one of the most stable and least soluble material. 5 It possesses excellent osteoconductivity or bone bonding ability and bioactivity because the chemical composition is similar to that of natural bones and teeth.6,7 However, due to its poor mechanical properties, 8 use of the material is restricted for load-bearing clinical applications.9–11 Composite of polymer and ceramic might exhibit a great role in bone regeneration by eliminating some weakness of each group to deliver a better quality and interface attachment for the new bone tissue. 12
Previously, several technologies have been reported to fabricate PLA/HA composite such as solvent casting, 13 electrospinning, surface coating, extrusion, compounding and forging,14–16 etc. In the current study, a PLA/HA composite was developed using injection molding which is similar to extrusion method, but the difference is screw operation. The screw of injection molding machine not only rotates continuously but also moves forward and backward in relation to the steps of the molding cycle. Furthermore, the mold is equipped with a cooling system to control cooling and solidification of the material. Industrially, injection molding is therefore highly productive and providing high accuracy. 17 With the intention of being use as a bone fixation, the tensile strength and compressive strength of the PLA/HA composite should be in the range of 50–150 MPa and 130–180 MPa, respectively, which are matched with those cortical bone.18,19 One of the most widely used manufacturing processes is an injection molding because the variety of materials and complexity of components could be produced. Not only several fabricating techniques used to improve mechanical properties of the composite, but grafting of the polymer was also reported in literatures.20,21 Polymer grafting could establish direct chemical bonding between polymers and HA by inducing the reactive hydroxyl groups onto the surface of HA particle resulting in an improvement of mechanical strength. 22
In this work, 70/30 weight ratio of PLA/HA composite was fabricated by melt extrusion followed by injection molding process. In order to improve mechanical properties of PLA/HA composite, glycidyl methacrylate (GMA), the most widely used reactive compatibilizer,22–25 was introduced into the PLA/HA composite as a grafting side chain. PLA/HA composite with different GMA contents, 1, 3 and 5 wt.% was fabricated to investigate theirs effect on the properties of PLA/HA composite. In addition, Joncryl containing nine glycidyl methacrylate functions as a chain extension/branching agent21,26–28 was also explored. Fixed amount of DCP as an initiator was also added during the compounding process to enhance the grafting condition. The structural characterization and the mechanical performance of these various composites prepared by injection molding were then evaluated as a function of GMA/Joncryl content. The cell viability on the surfaces of the specimens was also investigated by Alamar Blue assay and alkaline phosphatase (ALP) expression. Furthermore, the cell morphology on the surfaces of the specimens was determined by an SEM technique. Expectantly, the studied composite could be used as internal bone fixation devices such as screws, pins and plates.
Experimental
Materials
Pure hydroxyapatite used in this study was prepared through a mechanochemical method.
29
Mixture powders of calcium carbonate (CaCO3) and calcium phosphate dibasic (CaHPO4) at molar ratio 1.5 were reacted under the chemical reaction given in the following equation

All raw materials were used as received. CaCO3 (Carlo Erba Reagenti, Italy) and CaHPO4 (98.0–105.0%: Sigma-Aldrich, Germany) were ball-milled with zirconia balls in the presence of deionized water as a medium for 48 h, before drying to obtain an uncalcined HA. By chemical analysis, Ca/P was found to be 1.69 which was close to pure hydroxyapatite (1.67). Uncalcined HA in combination with PLA was reported as a good resorbability (induced degradation rate after implantation), biocompatibility and excellent remineralization ability. 13 Therefore, uncalcined HA was chosen to be used as a starting material in this study. The obtained powders were grounded and sieved to attain the average particle size (D0.5) about 2.5 µm particle size in the range of 0.5–9.5 µm. Characterization of HA powders was done, using X-ray diffraction (XRD) (JEOL JDX 3530) with CuKα source (Kα = 1.5406 nm) operating at 30 mA and 50 kV. XRD measurement was conducted at 20–60° 2θ, scan speed of 2°/min and a step size of 0.02°. The XRD spectra were analyzed using JADE software and ICDD cards. Particle size analysis was measured by a laser diffraction technique (Mastersizer 2000, Malvern instruments, UK).
The PLA with weight average molecular weight about 120 KDa (PLA 4043D) used in this study was purchased from Nature work Co. (USA) having the glass transition (Tg) and melting point (Tm) at around 71℃ and 156℃, respectively. PLA pellets were vacuum dried at 60℃ for 24 h to remove residual moisture before mixed with other components such as HA, glycidyl methacrylate (GMA, C7H10O3, 97%: St Louis, MO, USA) and dicumyl peroxide (DCP, C6H5C(CH3)2]2O2, ≥97%: St Louis, MO, USA). GMA and DCP supplied by Sigma-Aldrich Co. LLC were used as received. In this study, DCP was chosen as an initiator for grafting reaction of GMA onto PLA main chain due to a suitable decomposition temperature at the blending condition. Joncryl ADR®-4368 (the styrene acrylic multi-functional oligomeric chain extender: MW ∼6800 g/mol) was obtained from BASF (Thai) Limited (Thailand).
Fabrication of PLA/HA composite
Different formulations of the PLA/HA composites were prepared via the twin screw extruder (Labtech LTE-20-32, Labtech Engineering, Thailand) at 175℃ to obtain compounding pellets. Then the compounding pellets were fabricated through the injecting molding (Niigata MD30S-III, Japan) to produce flexural strength specimens according to ASTM D790. 30 The barrel temperature was set from 167 to 185℃ with an injection speed of 7.6 cm3/s and holding pressure range between 90 and 110 MPa.
Characterization and testing
Differential scanning calorimetry (DSC822e Mettler Toledo, Switzerland) was used to study the thermal properties. The samples were scanned twice from −60 to 250℃ with a heating and cooling rate of 20℃/min under a nitrogen purge. Gel permeation chromatography was used to determine the molecular weight of the starting materials. Morphology of the samples was analyzed by using a scanning electron microscope (SEM: Hitachi FE8030, Japan) equipped with an Energy Dispersive X-ray Spectrometer (EDS) on the fractured surface of the samples after gold coating to prevent over charging and to improve conductivity. NMR spectra were recorded at a frequency of 500 MHz on AVANCEIII 500 MHz digital NMR spectrometer (Bruker Biospin; AV-500, Germany) using CDCl3 as a solvent. Attenuated total reflectance (ATR-FTIR) spectra were recorded on Nicolet model iS5 (USA) spectrometerat a resolution of 4 cm−1. For mechanical properties testing, 3-point bending was performed on at least five specimens of each formulation (4.8 mm width × 57 mm long × 4.0 mm thickness) by a universal testing machine (Instron 55R4502, USA) with 10 kN load cells at a cross head speed of 1 mm/min. Averages values and standard deviations were reported. The melt flow index (MFI) of the samples was determined using a Melt Flow Indexer, MFI 10, Davenport (Lloyd Instruments, UK). The temperature of the melt was kept at 175℃ and a 2.16 kg load was applied to extrude the molten sample. The MFI was averaged from at least five determinations. Five samples of each formulation were used to measure the density by a gas pycnometer (Ultrapyc 1200e, Quantachrome Instrument, USA) under gas Helium atmosphere.
In vitro cell proliferation and differentiation
Cell culture
The MC3T3-E1 subclone 4 mouse pre-osteoblast cell line (ATCC CRL-2593) was purchased from the American Type Culture Collection (ATCC) and routinely cultured in alpha minimum essential medium (α-MEM, Gibco) supplemented with 10% fetal bovine serum (FBS, Gibco) and penicillin/streptomycin (1X, Gibco), and maintained at 37℃ in a humidified incubator with 5% CO2.
Cell proliferation
When the confluency reached 80%, the cells were detached using 0.25% Trypsin/1 mM EDTA-4Na and directly seeded at a density of 5 × 103 cells cm−2 and a volume of 25 µL to completely cover only the surface of square-shaped specimens (5 mm × 5 mm). After allowing initial cell adhesion at 37℃ in a humidified incubator with 5% CO2 for 2 h., the unattached and loosely attached cells were removed by gentle washing with phosphate buffered saline (PBS, pH 7.4). The specimens were transferred to a 96-well cell culture plate and incubated in 150 µL fresh completed culture media for up to 10 days at 37℃ with 5% CO2. Cell viability was assessed by Alamar Blue assay which quantifies the number of metabolically active cells. After allowing cells to grow for a given time, the culture medium was replaced with 250 µL of a 0.001% w/v of Alamar blue (Sigma) in serum-free culture media. After 4 h of incubation at 37℃ and 5% CO2, the reduction of Alamar Blue in culture supernatants was determined using fluorescent plate reader (Perkin Elmer’s Wallac 1420 Multilabel Counter) for excitation at 530 nm and emission at 590 nm. The same procedure was repeated with the samples without cells as a blank. Tissue culture-treated polystyrene petridish (TCPS) was used as a positive control. Experiment were run in triplicate (n = 3).
Cell morphology
The morphology of the attached cells was observed by a Hitachi SU8030 SEM. Cells were fixed with 4% paraformaldehyde in PBS, sequentially dehydrated with a graded series of ethanol solutions (30–100%) and dried in a Emitech K850 critical point dryer (Emitech Ltd, UK). The samples were subjected to gold sputtering to prevent over charging and improve conductivity and then transferred to the microscope where they were observed at an accelerating voltage of 15 kV.
Cell differentiation
The MC3T3-E1 cells were seeded on the composite samples using the same procedure as in Cell proliferation section. After transfering the samples to a 96-well cell culture plate, cells were incubated in 150-µL osteogenic media consisting of α-MEM with 10% FBS, 0.1 µM dexamethasone (Sigma), 10 mM beta-glycerol phosphate (Sigma, USA) and 50 µg/mL ascorbic acid-2-phosphate (Sigma) for a given time of 14 days and 21 days at 37℃ with 5% CO2. After washing with PBS, cells were frozen and thawed five times, sonicated in 150-µL lysis buffer (0.1 M glycine (pH 9.6) containing 1 mM MgCl2, 1 mM ZnCl2 and 1% Nondet P-40) and centrifuged. Aliquots (30 µL) of the supernatant were added to a 96-well plate for the determination of ALP activity by measuring the release of p-nitrophenol from p-nitrophenylphosphate. The p-nitrophenyl phosphate solution (90 µL) was added and incubated at 37℃ for 30 min. The reaction was terminated with 12.5 µl of 3 M NaOH, and absorbance at 405 nm was determined using a microplate reader (Bio-Tek ELx808). PBS was applied as a control. Each experiment was carried out in triplicate. Enzymatic activities were normalized to total protein content as determined by a bicinchoninic acid assay (Novagen) with bovine serum albumin (BSA) as the standard. ALP activity was expressed as nmol/mg protein/assay time.
Cell mineralization
The MC3T3-E1 cells were grown on the composite samples for 14 and 21 days using the same procedure as in Cell proliferation section. Mineralization of adherent MC3T3-E1 cells was then quantified using an Alizarin red-based assay as previously reported. 31 Briefly, cells were fixed in 10% (v/v) paraformaldehyde for 15 min, prior to staining calcium deposits using 120 µL of Alizarin Red S. (pH 4.1) for 20 min. Stained samples were extensively washed with deionized water to remove the unincorporated dye. Acetic acid (10% v/v, 150 µL) was added to each well and the plate was shaken for 30 min at room temperature. The still adherent cells were scraped from the plate was scraped with a sterile pipette tip and transferred to a microcentrifuge tube. Cell suspension was heated to exactly 85℃ for 10 min and transferred to ice until completely cooled. After centrifugation, supernatant was removed to another microcentrifuge tube and 25 µL of 10% (v/v) ammonium hydroxide was added to neutralize the acid. Absorbance at 405 nm was determined using a microplate reader (Bio-Tek ELx808).
Statistical analysis
All data were expressed as mean ±standard deviation (SD). Data analysis was performed using the SPSS statistical software package for Windows version 12.0 (SPSS Inc., USA). Statistically significant differences (p < 0.05) between multiple means were determined by the one-way ANOVA and the least significant difference (LSD) test. The independent samples t-test was used to compare the difference of the two populations.
Results and discussion
Compounding and characterization of PLA/HA composite
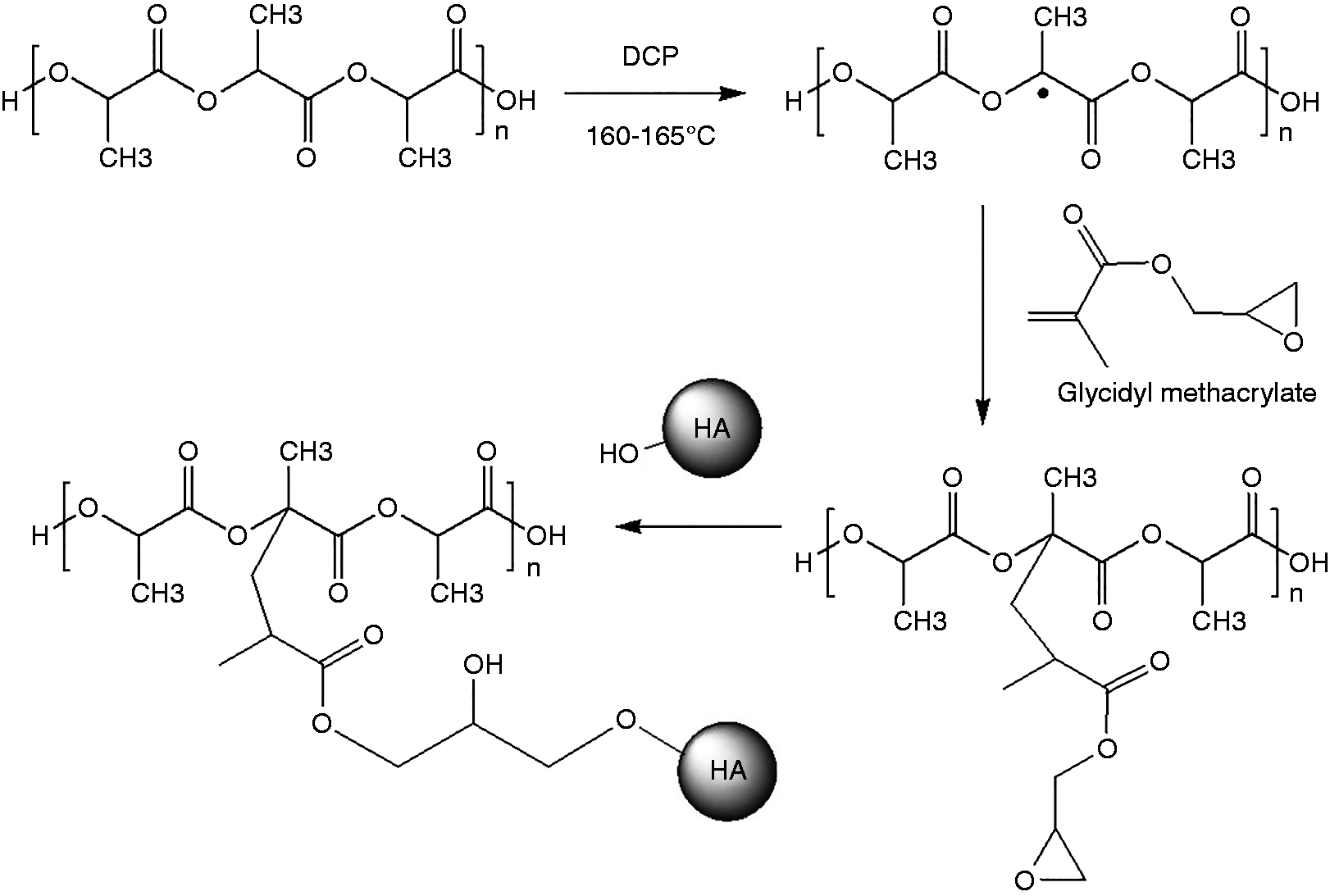
A series of PLA/HA composite with various blending amounts of GMA was compounded using the twin screw extruder. In addition, various amounts of GMA/Joncryl as well as DCP (10 wt% GMA) were also added as a reactive compatibilizer and initiator, respectively. Chemical structure of the composite was evaluated via FTIR and NMR spectroscopy. The proposed reaction among PLA, GMA and HA was showed in Scheme 1. Without DCP initiator, GMA can possibly use epoxide group to graft onto PLA either at the chain ends or backbone and leaving acrylate group as a side chain.21,32 However, with DCP, not only epoxide group of GMA was reacted but also methacrylate group of GMA was undoubtedly grafted onto PLA macro-initiator and leaving epoxide group as a functional side chain group.33–36 After mixing with HA, only epoxide side chain group could possibly react with hydroxyl group of HA through ring opening reaction.
FTIR spectra, in the region of 1300–1900 cm−1 of PLA/GMA5/HA composite with or without DCP and their subtraction result, are shown in Figure 1. Characteristic bands at 1760 and 1720 cm−1 are due to the C = O stretching modes of PLA and GMA, whereas bands at 1640 cm−1 were assigned to C = C stretching modes of GMA.
37
A unique band at 815 cm−1 is due to the =CH2 deformation mode of GMA. The band ratio of those two C = O stretching mode can be employed to determine the GMA content in the composite. However, the separated bands at 1640 and 815 cm−1 are more convenient for determination of C = C content of GMA. The subtraction spectrum of the PLA/GMA5/HA composite without DCP from that of with DCP shows the negative result at 1720 and 1640 cm−1 meaning that there is less double bond content of GMA in the composite with DCP. In other words, GMA has more chance to react with PLA in the case of the composite with DCP,
38
either using epoxide group or acrylate group, leading to more reduction of the C = C band intensity. On the other hand, the composite without DCP can only use epoxide group to react with PLA leaving more intensity of C = C bond. This implies that the grafting reaction in the composite with DCP is more efficient than that of without DCP.
(a) spectra of (a) PLA, (b) PLA/GMA5/HA, (c) PLA/GMA5/DCP/HA and (d) the subtraction result of (c)–(b). Proposed chemical reactions among PLA, GMA and HA.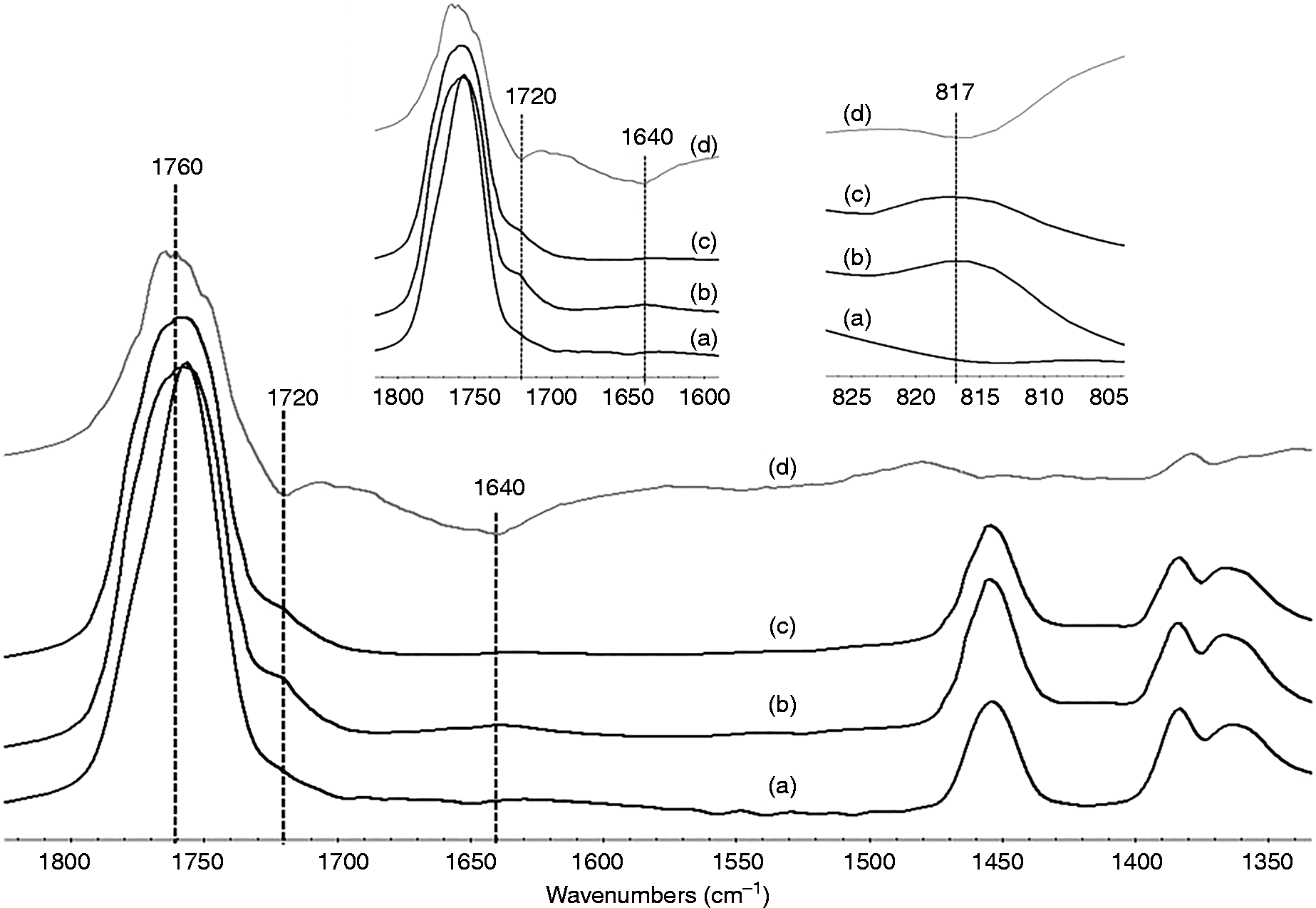
In addition, the decrease of the intensity of the –CH2 deformation in the composite with DCP at 815 cm−1 also confirmed the higher grafting efficiency of GMA as compared to that of the composite without DCP. Figure 2 shows FTIR spectra of PLA/GMA5 and PLA/GMA5/HA composite compared with PLA. Characteristic band at 920 cm−1 belongs to epoxide group. As epoxide group reacts with HA in the composite, the intensity of epoxide band at 920 cm−1 was reduced. This implies the chemical bonding between GMA and HA.
FTIR spectra of (a) PLA, (b) PLA/GMA5 with DCP, and (c) PLA/GMA5/DCP/HA composite.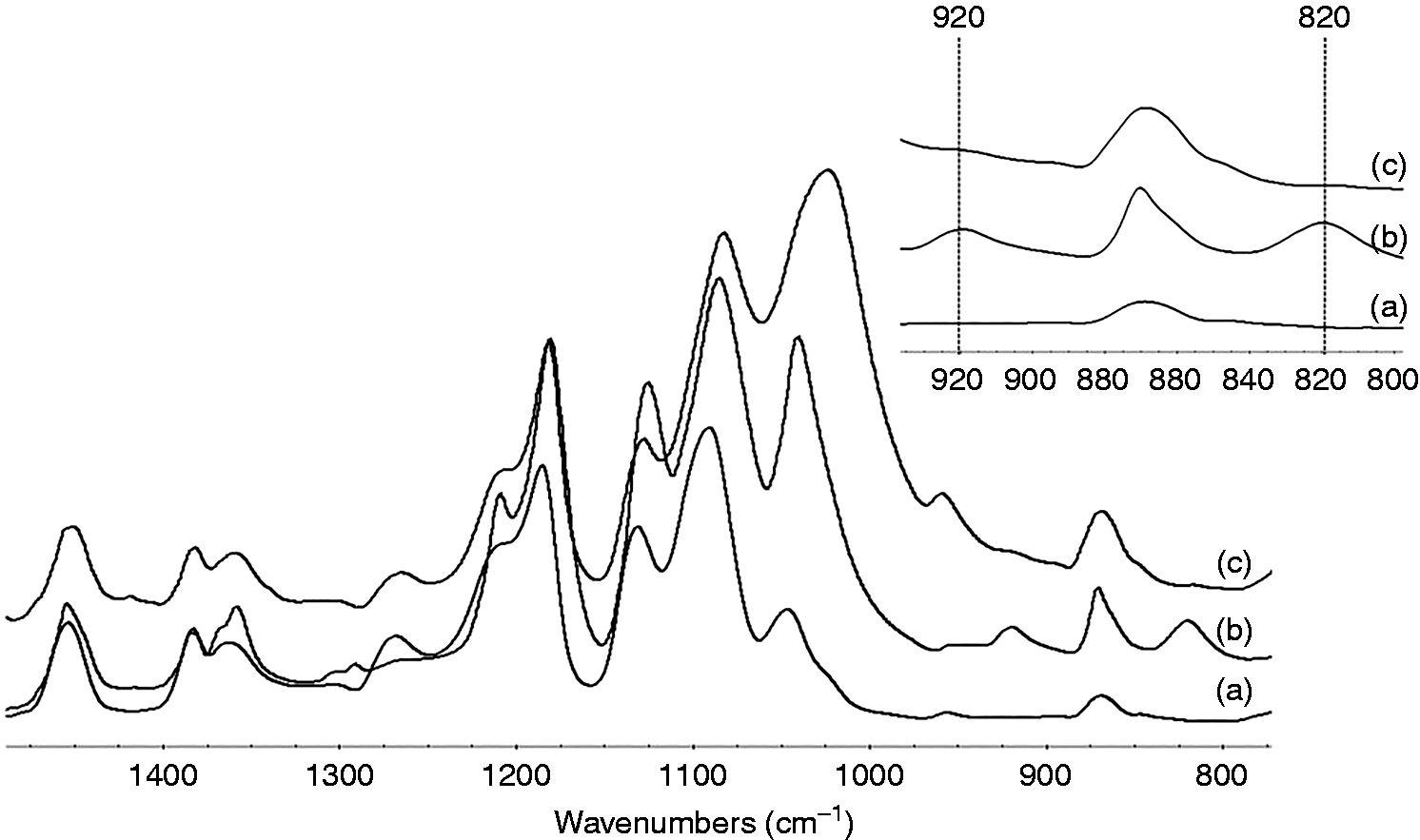
Figure 3 compares NMR results of the PLA/GMA5/HA and PLA/GMA5/DCP/HA composite. Apart from chemical shifts belonging to PLA at 1.5 and 5.1 ppm, new chemical shift at 5.6 and 6.2 ppm corresponding to the hydrogen next to double bond of GMA was observed in both the composites. However, the composite with DCP shows a lower integration at 5.6 and 6.2 ppm than that of the composite without DCP. This implies that double bond of GMA has more chance to react with PLA (grafting) leading to lower integration.
NMR spectra of (a) PLA, (b) PLA/GMA5/HA and (c) PLA/GMA5/DCP/HA.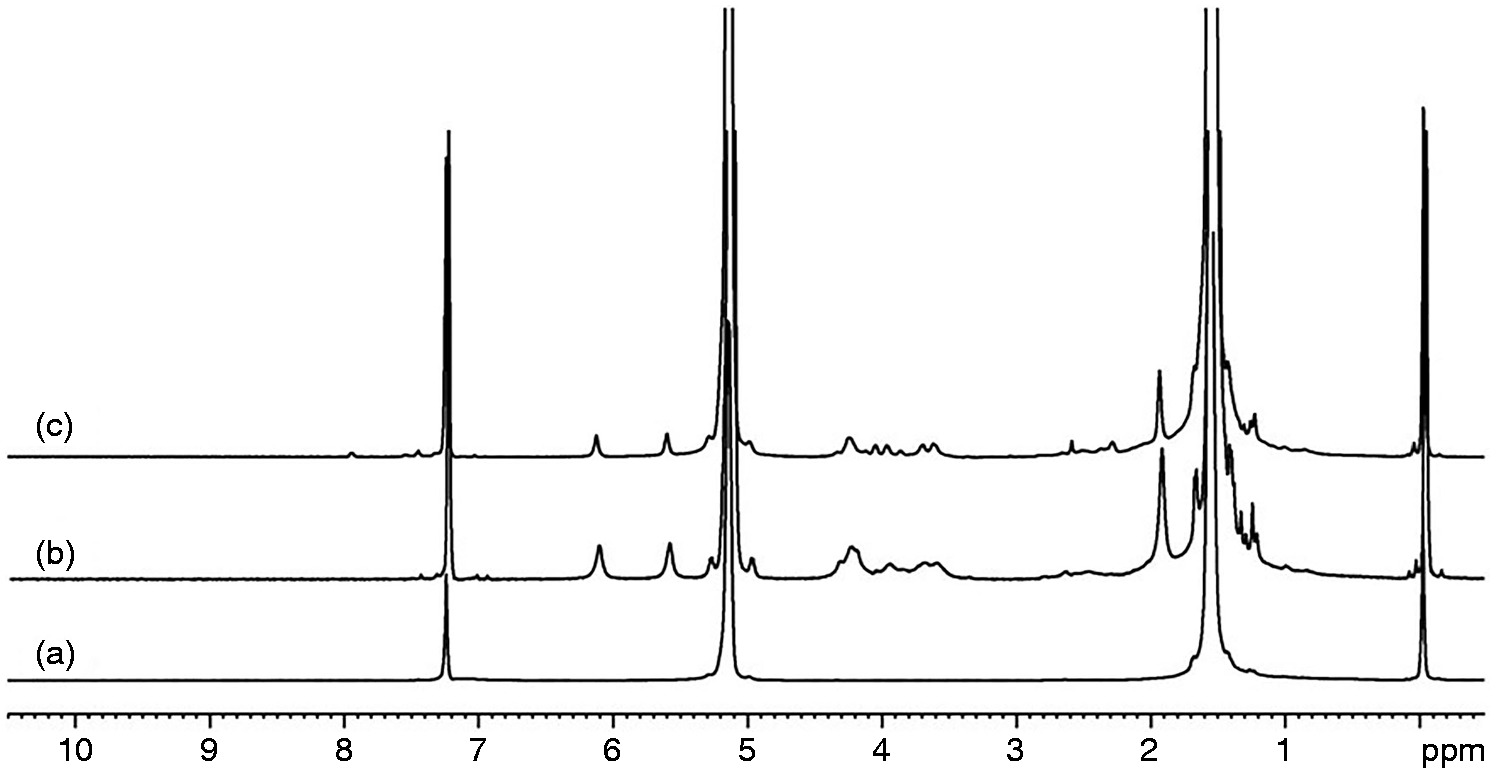
Mechanical properties of PLA/HA composite
Flexural strength of PLA/HA composites.
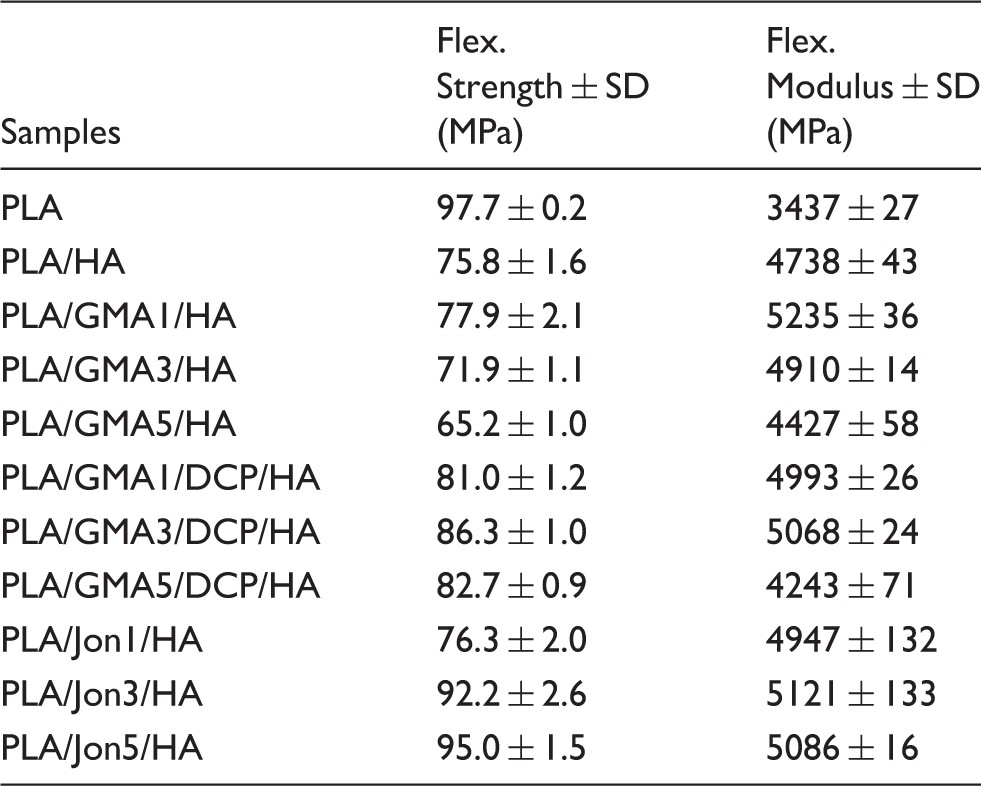
To improve the compatibility between HA and PLA, various amount of GMA/Joncryl and DCP were introduced as reactive compatibilizer/chain extender and initiator, respectively. It was found that the flexural strengths of PLA/GMA/HA composites with DCP were obviously increased to 86 MPa. This was probably because GMA acts as chemical linkage between PLA and HA, as revealed by NMR and FTIR, and enhances the compatibility of PLA and HA resulting in higher flexural strength. By adding Joncryl 1 wt.%, the strength was even lower than that of adding GMA 1 wt.% with DCP. However, better result was obtained when adding higher contents of Joncryl as a chain extender in the composite where higher contents of epoxide groups are available for reacting with HA. Thus, higher flexural strength was obtained, especially at 5 wt.% Joncryl.
Generally, in PLA/HA composite, PLA is denoted as matrix, which is responsible for supporting the filler by maintaining their relative positions. And HA is referred to filler which induces the physical and biological properties of the matrix. 39 To improve the mechanical properties of the PLA/HA composite, HA acts as a reinforcing agent to meet performance criteria for a particular application such as implant.40,41 For biocompatibility and bioactivity of the PLA/HA composite,42–46 HA could significantly promote cell proliferation and tissue regeneration42–45 and an apatite layer could be formed on the surfaces of PLA/HA composite after soaking in simulated body fluid 46 which will need further investigation in the near future particularly for this system. However, the several results of using PLA-CaP composites in vitro and in vivo have reported the increase of the biocompatibility and bone bonding ability as compared to the controlled PLA group.40,47–49 These results suggest that PLA-CaP composites have superior potential for clinical applications.
Melt flow index (MFI) and density of PLA/HA composites.
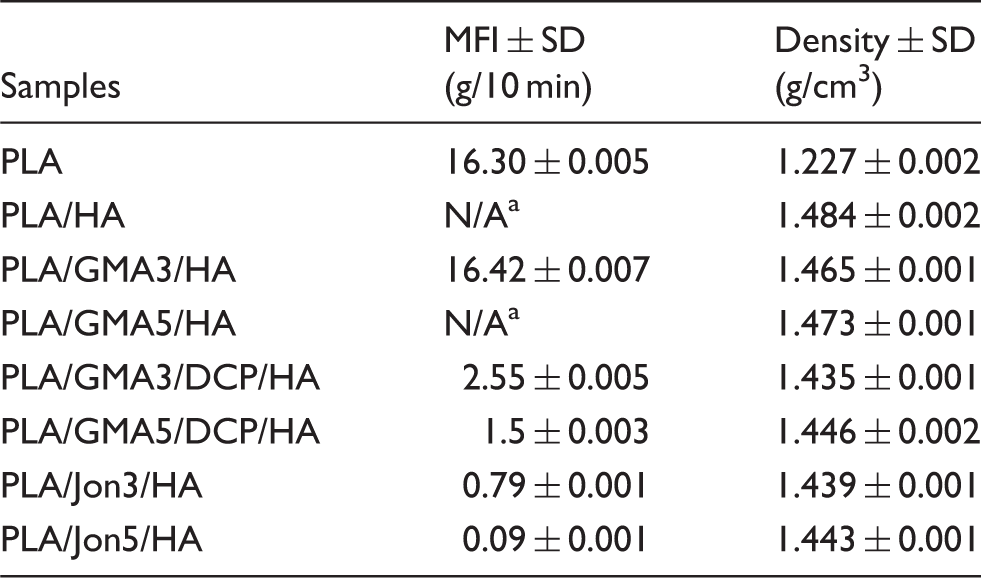
aN/A = MFI is too low to detect using the same load at 2.16 kg.
Scanning electron microscope
The information about the distribution of inorganic filler in PLA matrix could present by morphology of the composite samples. Scanning electron microscope (SEM) images coupled with EDS traces in Figure 4 reveals the HA particles uniformly dispersed in the composites. The agglomeration of HA particles could be found in some part of the composites and some parts were left with voids due to the filler detachment showing the distribution of inorganic was homogeneous through mix processing. However, the appearing of the voids suggested that the interface between PLA and HA filler required to be improved, because the voids may cause initial failure of the composite, resulting of limiting the mechanical properties of PLA/HA composite. Essentially, a good distribution of the inorganic filler in the PLA matrix is necessary during melt compounding.
14
Morphology and EDS traces of (a) Pure PLA, (b) PLA/HA (c) PLA/GMA1/HA, (d) PLA/GMA3/HA and (e) PLA/GMA5/HA.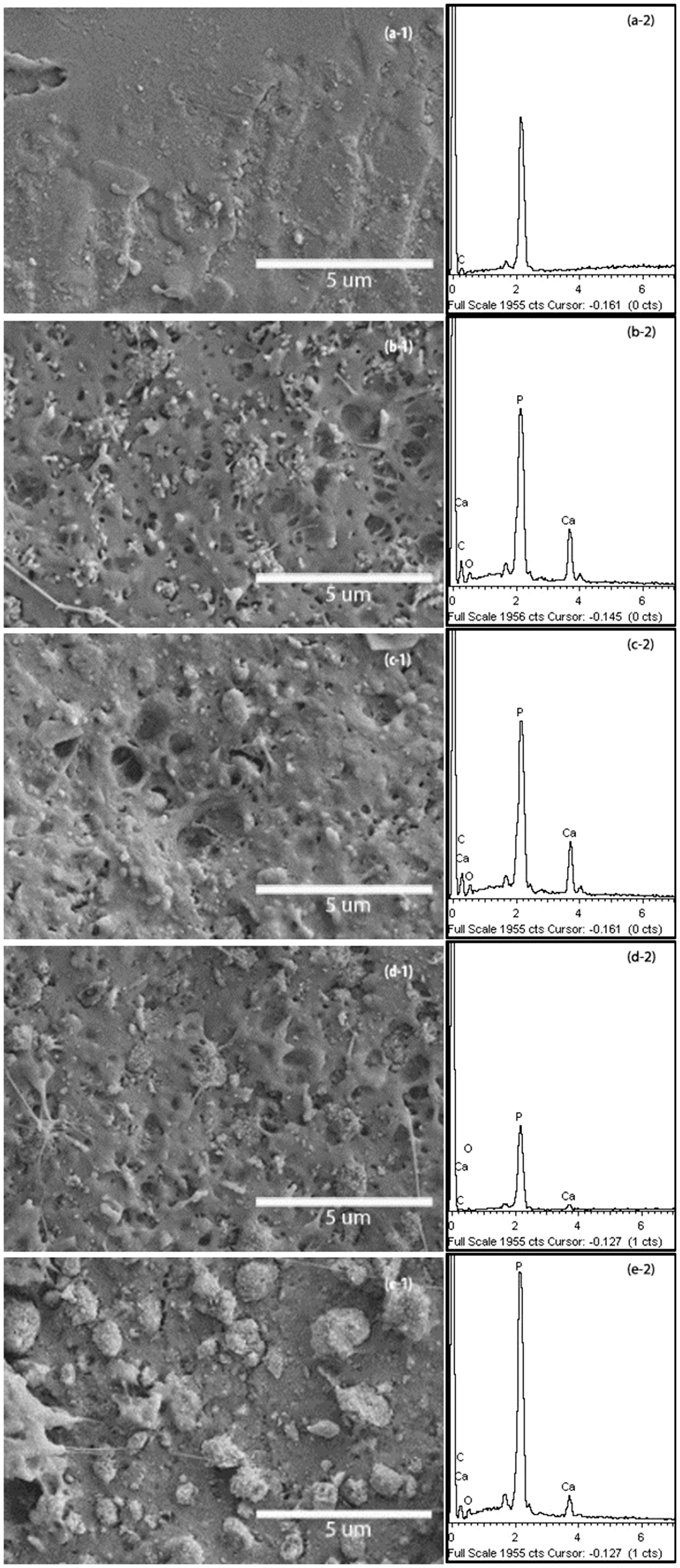
Figure 5 shows SEM micrographs of fractured surface of PLA/GMA/DCP/HA and PLA/Jon/HA composites. They show the similar morphologies to those of the samples without DCP. However, the long fibrils of PLA could observe in PLA/GMA3/DCP/HA sample. These long fibrils could be responsible for the strength and the elastic of the composite. The significant morphology change could be seen in samples mixed with Joncryl. The uniform agglomeration of HA particles oriented in PLA matrix coupled with micropores and small voids particularly in PLA/Jon5/HA. By introducing a compatibilizer such as Joncryl could prevent the agglomeration of the filler particles.
18
Morphology of (a) PLA/GMA1/DCP/HA, (b) PLA/GMA3/DCP/HA, (c) PLA/GMA5/DCP/HA, (d) PLA/Jon1/HA (e) PLA/Jon3/HA and (f) PLA/Jon5/HA.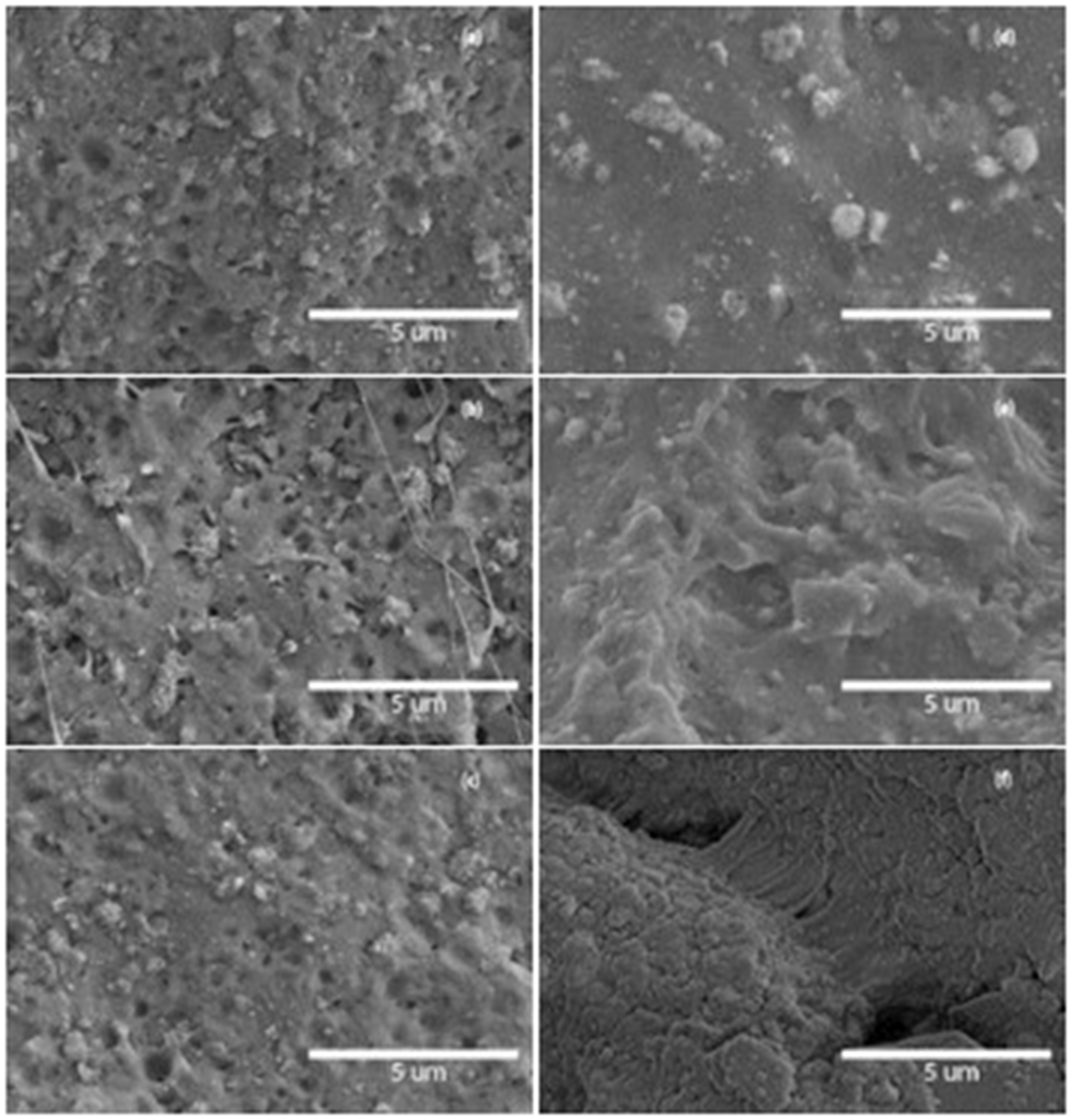
Cell proliferation and differentiation
Resazurin reduction assay was performed to evaluate mouse osteoblastic MC3T3-E1 cell proliferation for 10 days on a pure PLA and 11 different PLA/HA composites, and the TCPS plates were used as control (Figure 6). Due to quantitative differences of surface areas between tested samples (0.25 cm2) and the control (0.32 cm2), the measured fluorescence intensity was normalized by surface area. All tested samples, with the unexpected exception of the PLA/GMA3/HA composite, supported significantly lower cell proliferation than the TCPS control (day 3 to day 10; p < 0.002). There were no statistically significant differences (p > 0.05) in the normalized numbers of attached MC3T3-E1 cells on the PLA/GMA3/HA and the TCPS surfaces during the culture times of three day and five days. However, unlike TCPS, little further proliferation on the PLA/GMA3/HA occurred beyond day 5.
Mouse osteoblastic MC3T3-E1 cell proliferation on different PLA/HA composites. Error bars reflect SD and are not visible if the SD is smaller than the symbol (n = 3).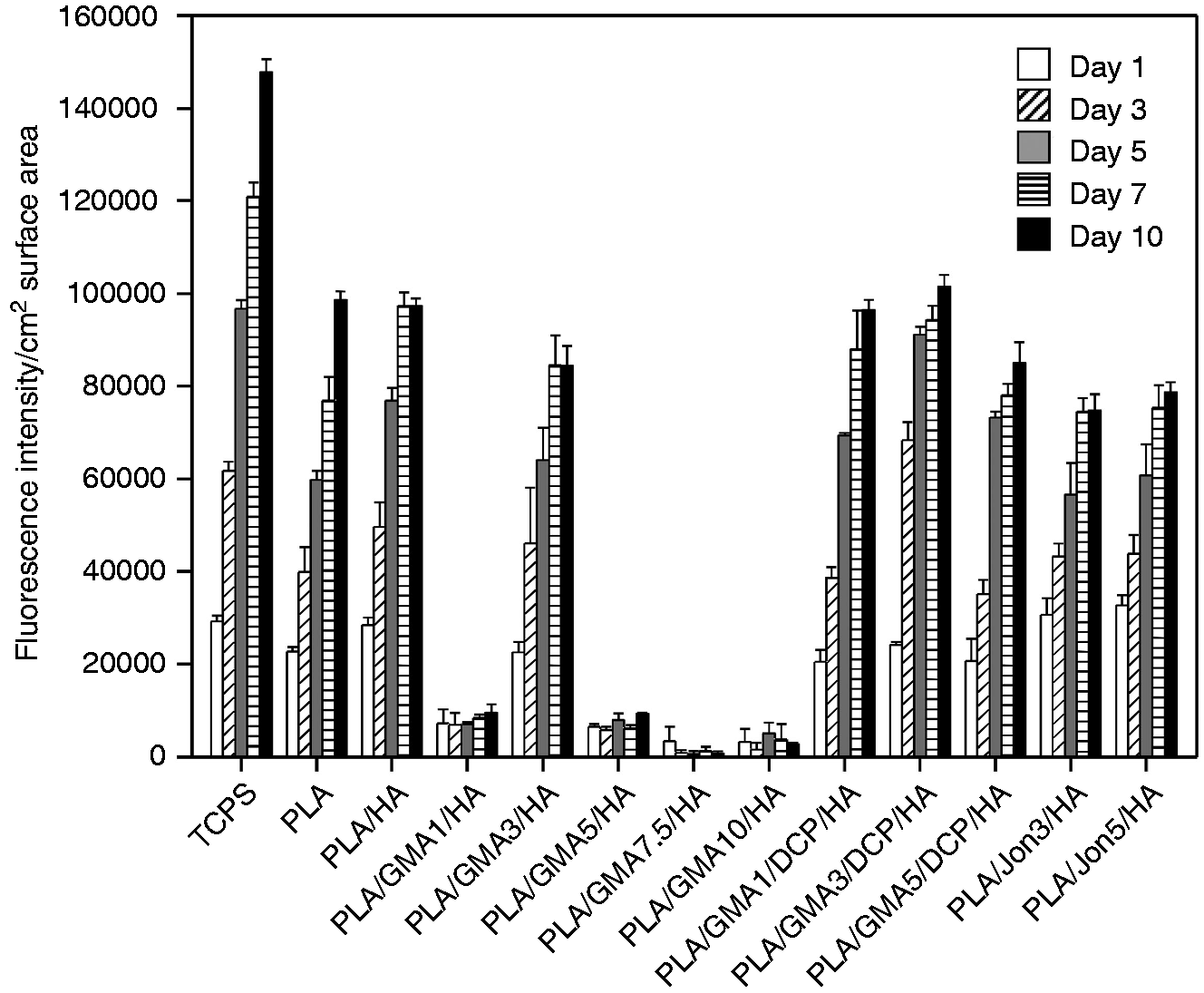
For clarity in comparison, the proliferation data for only GMA-bearing, GMA/DCP-bearing and Joncryl-bearing composites in Figure 6 are separately replotted as a function of incubation time in Figure 7(a) to (c), respectively. Osteoblast seeding resulted in initial attachment and subsequent proliferation for all tested GMA/DCP-bearing and Joncryl-bearing surfaces. Addition of only GMA monomer to the PLA/HA composite (DCP-free samples) was found to exhibit the strongly negative effect on the MC3T3-E1 cell proliferation (called non-growth substrates), with the unexpected exception of the PLA/GMA3/HA composite (Figure 7(a)). Relatively lower number of cells (0.11- to 0.25-fold), as compared to PLA/HA, initially adhered to these PLA/GMA/HA surfaces with no further increase after 10 days of incubation. Figure 8 shows some examples of semi-log scale plots from the same data in Figure 6. It was found that MC3T3-E1 cells on all tested surface, with the exception of PLA/GMA3/DCP/HA and non-growth substrates, exhibited exponential growth (constant growth rate) during the first five days or longer. The exponential growth period (day 1–3) of PLA/GMA3/DCP/HA was shorter than the others.
Proliferation data for (a) GMA-bearing, (b) GMA/DCP-bearing and (c) Joncryl-bearing composites as a function of incubation time. Error bars reflect SD and are not visible if the SD is smaller than the symbol (n = 3). Proliferation data on some tested material surfaces plotted on a semi-log graph against incubation time. The dashed lines denote linear curve-fits of the exponential growth data where the growth rate is the slope of these linear curve.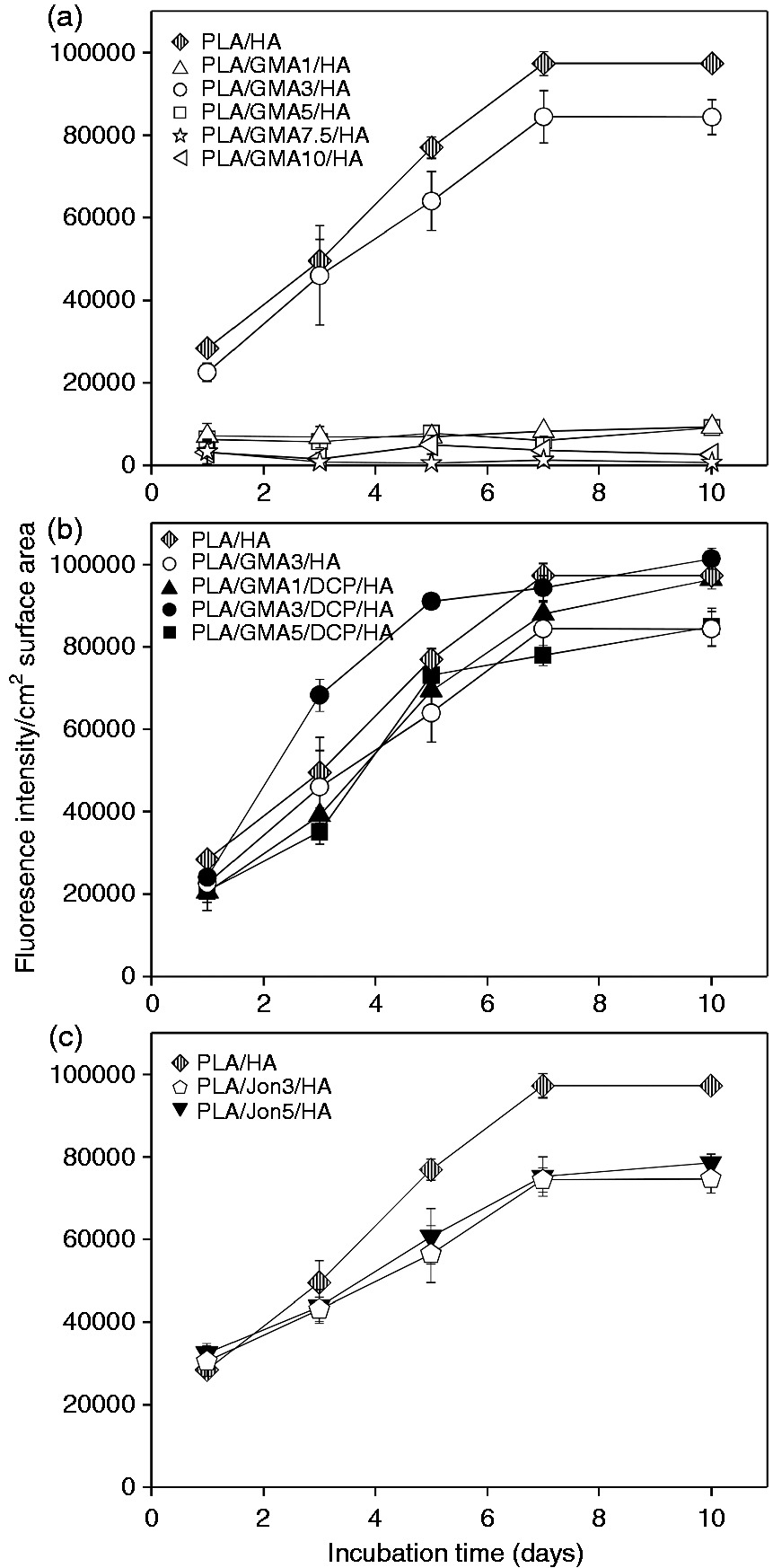
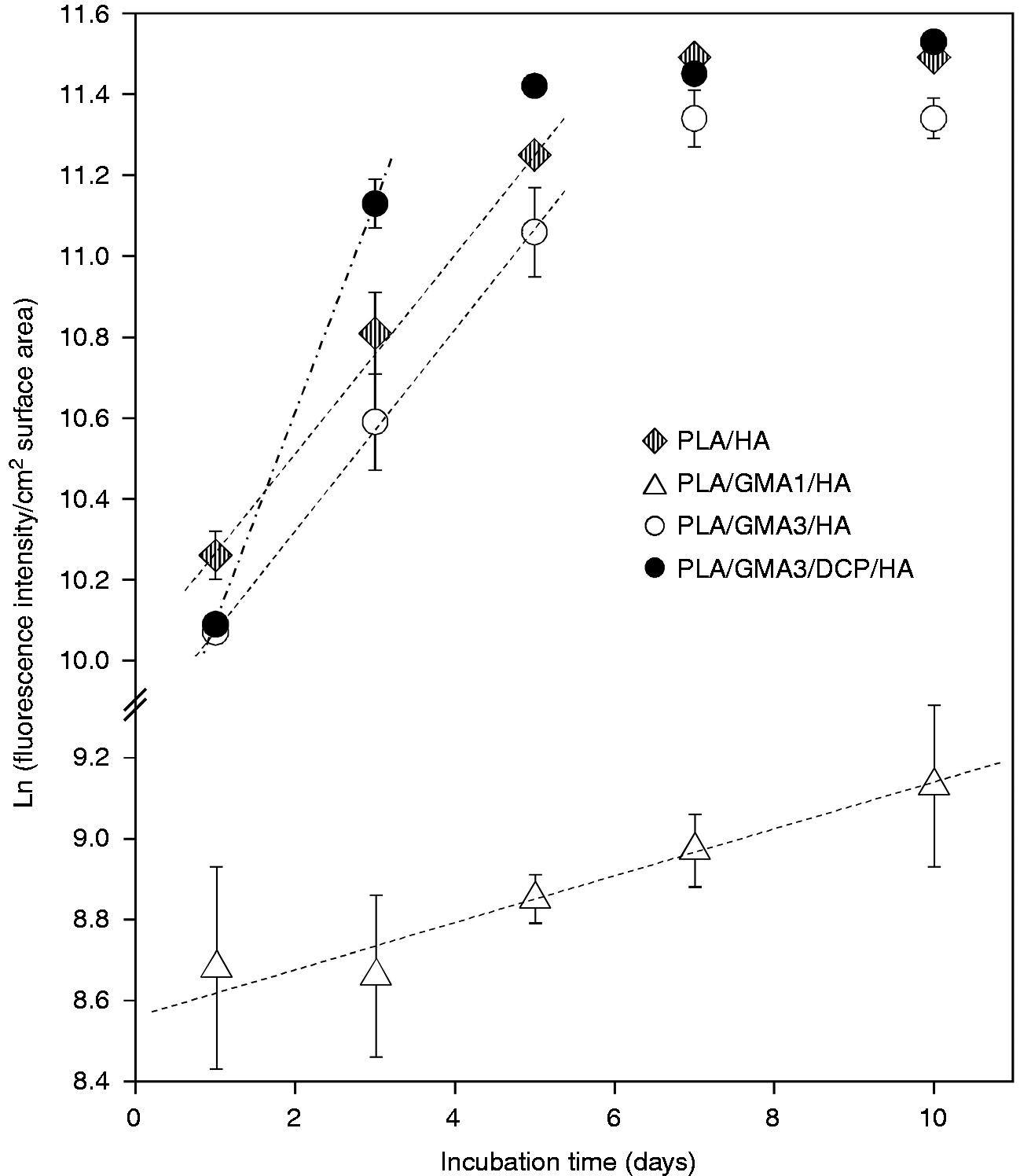
Growth rate of MC3T3-E1 osteoblast cells on different PLA/HA composite and TCPS surfaces.
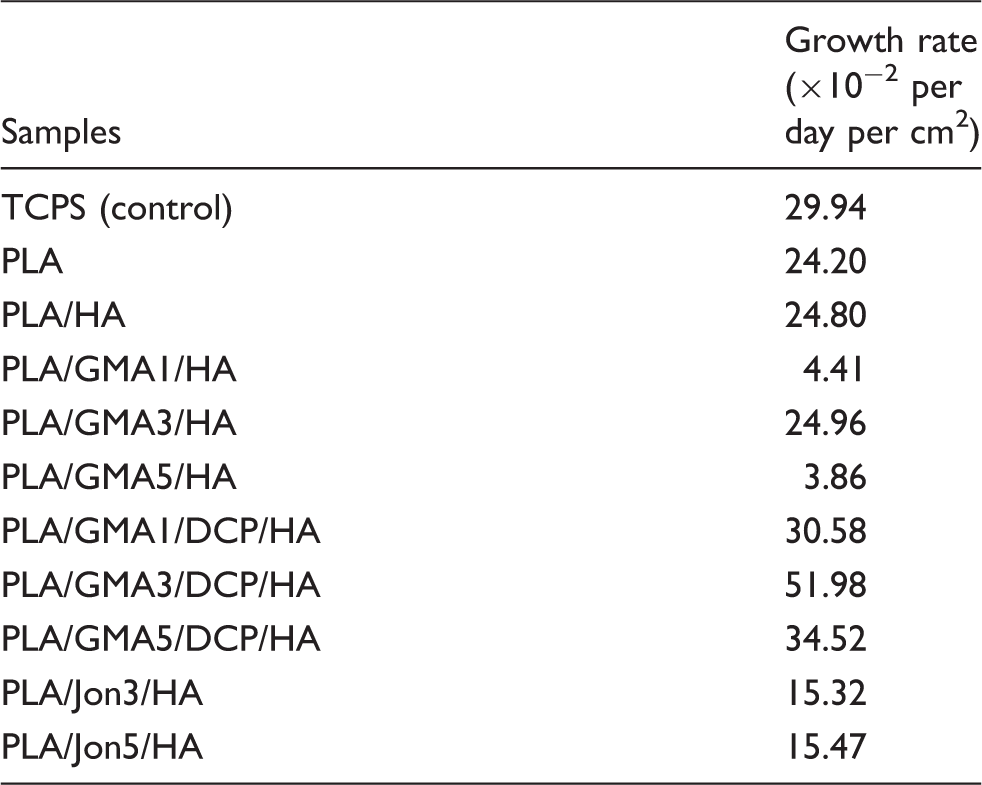
GMA is known to be concentration-dependent cytotoxic in vitro. 54 With overfeeding, a larger amount of unreacted GMA monomer remained from initiator-free preparation of the PLA/GMA/HA composite, as compared to DCP initiating polymerization of PLA/GMA/DCP/HA with the corresponding percentage of DCP. This was due to the fact that, without any initiator, each lactic acid backbone repeating unit of the PLA could react with at most one GMA molecule, while graft co-polymerization of GMA onto the PLA occurred in the presence of DCP initiator. Therefore, it is not unexpectedly to observe its inhibitory effect on MC3T3-E1 proliferation on the PLA/GMA/HA composites, with a growth rate close to zero (Table 2). This possibly indicated that remaining unreacted GMA was leached out in sufficiently amounts from the PLA/GMA/HA composite to locally harm cells. Note that all composite samples were pre-soaked in the culture media overnight prior to cell experiments to remove leachable substances. However, we unexpectedly found that a GMA concentration of 3 wt.% (Figures 6 to 8, Table 2) provided a weak promotive effect on MC3T3-E1 proliferation as seen in a slightly greater growth rate of PLA/GMA3/HA as compared to that of PLA/HA. Despite a significant lower adhered cell number at 24 h on the PLA/GMA3/HA surface (p < 0.001), the number of cells grown for 10 days became significantly greater as compared with PLA/HA (p < 0.001). This may contribute to physicochemical changes as a result of new formation of ester side groups on the PLA chain in quantitative sufficiency.
Polymerization of GMA monomers using DPC as an initiator appeared to not only solve the cytotoxicity problem of monomer leaching, but also possibly allow the polymerized GMA repeating units to effectively exhibit their cell growth supporting properties. It can be evidenced by the increases in growth rates of PLA/GMA3/HA and PLA/GMA5/HA by 2 and 9 folds, respectively, after the DPC initiator was applied (Table 3). PLA/GMA3/DCP/HA showed the highest growth rate of MC3T3-E1 osteoblast cells in this study. Moreover, PLA/GMA5/DCP/HA became a non-cytotoxic substance with a comparable growth rate to the PLA/HA composite. However, maximum cell numbers grown on all GMA-loaded composites were statistically less than those on the PLA/HA and the TCPS control.
MC3T3-E1 cells could initially adhere to PLA/Jon/HA composites at a comparable level to the PLA/HA0 (p > 0.05) and oligomeric chain extender Joncryl caused no harm to cells (Figures 6 and 7). However, Joncryl was found to reduce the growth rate (Table 3) and proliferation (Figure 7) of the PLA/HA composites in a concentration-independent manner. It was evident as that addition of Joncryl to the PLA/HA significantly decreased the attached cell numbers after seven days of cultivation (p < 0.001).
The morphology of the MC3T3-E1 cells cultured on all studied composite surfaces at day 3 and day 7 was examined by SEM (Figure 9). SEM revealed that PLA, PLA/HA, PLA/GMA3/HA, all of GMA/DCP-bearing and Joncryl-bearing composites supported MC3T3-E1 cell attachments and, at day 3, the cells were elongated and had spindle-like morphologies with good attachments to the surface. At day 10, the flatten cells on these substrates contacted each other and overlapped, and the surfaces were fully covered by a continuous sheet of MC3T3-E1 cells. In contrast, we observed relatively few cells with slightly elongated cells on all composite surfaces with poor cell supporting ability (PLA/GMA/HA with the exception of the PLA/GMA3/HA) and, even seven days post incubation, pseudopodia were rarely seen.
Morphology of MC3T3-E1 cells cultured on different PLA/HA composites examined by scanning electron microscopy.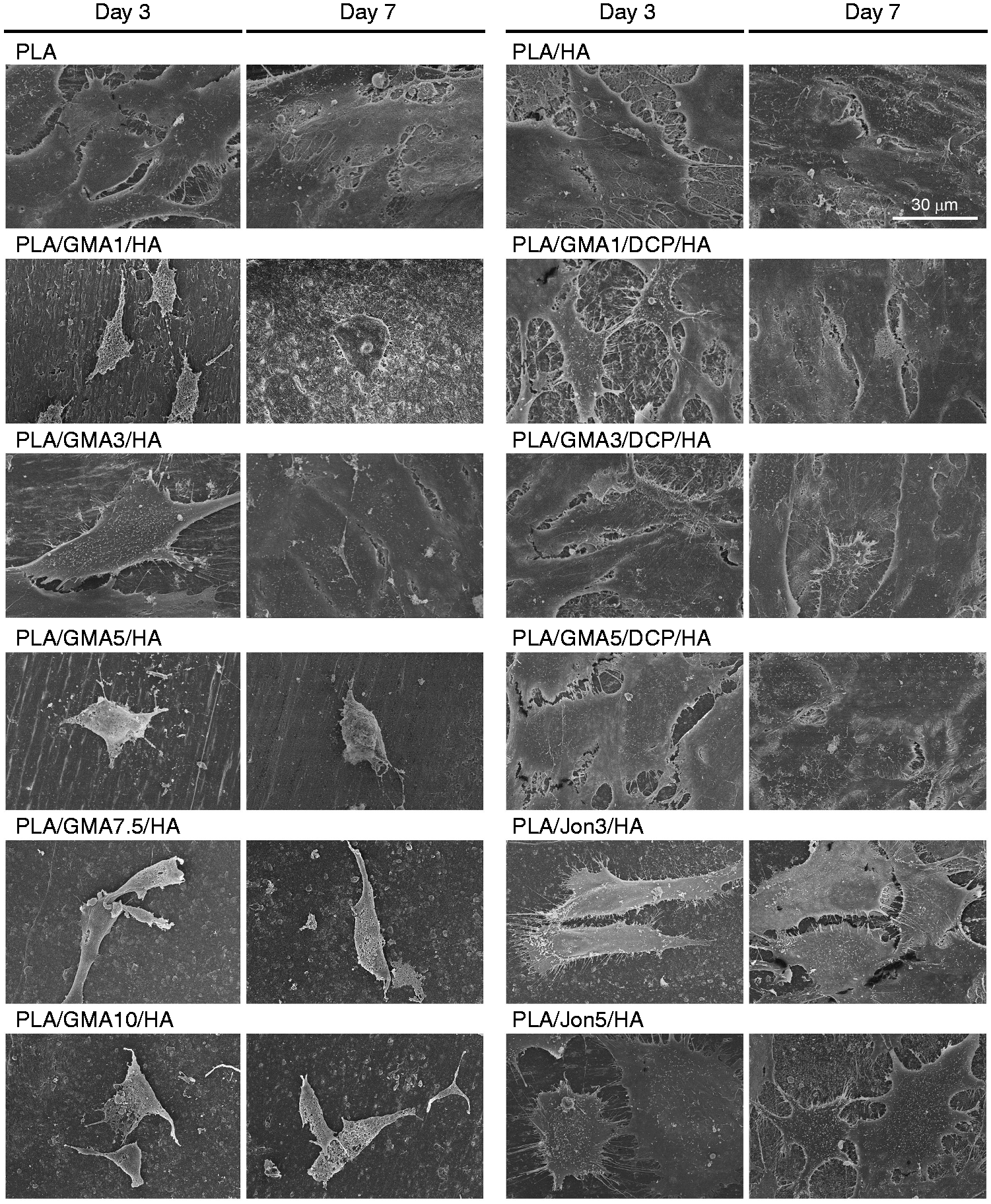
Alkaline phosphatase (ALP), an early differentiation marker for osteoblasts, is a membrane-bound enzyme believed to provide inorganic phosphate for mineralization. Some previously published studies found that MC3T3-E1 cells proliferated without ALP expression for the first period of culture, then underwent early osteoblastic differentiation as characterized by steady increases of ALP and subsequently declined.55–57 The time-dependent ALP expression of MC3T3-E1 cells was influenced by various factors such as differentiation stage, cell passage number, source of serum, and seeding density.57–59 To preliminarily evaluate the effect of GMA monomer, DCP initiator and Joncryl chain extension/branching agent on the differentiation of osteoblastic MC3T3-E1 cells, the activity of ALP at day 14 and day 21 of osteoblast proliferation and differentiation was examined (Figure 10). The experimental variables chosen in this study allowed MC3T3-E1 cells to highly express ALP on TCPS control and PLA at day 14 and significantly decreased at day 21 (p < 0.05). The ALP downregulation indicates a progress to the next stage in differentiation.
60
The substrate components could affect time-dependent differentiation as seen that the ALP activities of all HA-incorporated substrates remained constant after 14 days (p > 0.05). Addition of osteoconductive HA ceramic to the PLA induced ALP activity at day 14 (p = 0.177) and 21 (p < 0.001). A pronounced reduction in ALP activity was found for all tested PLA/GMA/HA composites as compared to their base material, PLA/HA (p < 0.001). The least reduction with an approximately two-fold decrease was observed for PLA/GMA3/HA at both tested incubation times. The DCP-initiated polymerization of the GMA monomers significantly elevated the ALP activity values of GMA-bearing composites (p < 0.001 as compared to the corresponding PLA/GMA/HA) back close to that of PLA/HA, especially for PLA/GMA3/DCP/HA and PLA/GMA5/DCP/HA (p > 0.2 as compared to PLA/HA). Moreover, ALP activity levels of the TCPS control were the greatest among all tested surfaces. Similar to GMA monomer, Joncryl chain extension/branching agent exhibited a moderate inhibitory effect on ALP activity of osteoblastic MC3T3-E1 cells (p < 0.001 at day 21 as compared to PLA/HA). However, we unexpectedly found more decrease in the ALP activity levels as less amount of Joncryl was added.
ALP activity of different PLA/HA composites. Error bars reflect SD and are not visible if the SD is smaller than the symbol (n = 3).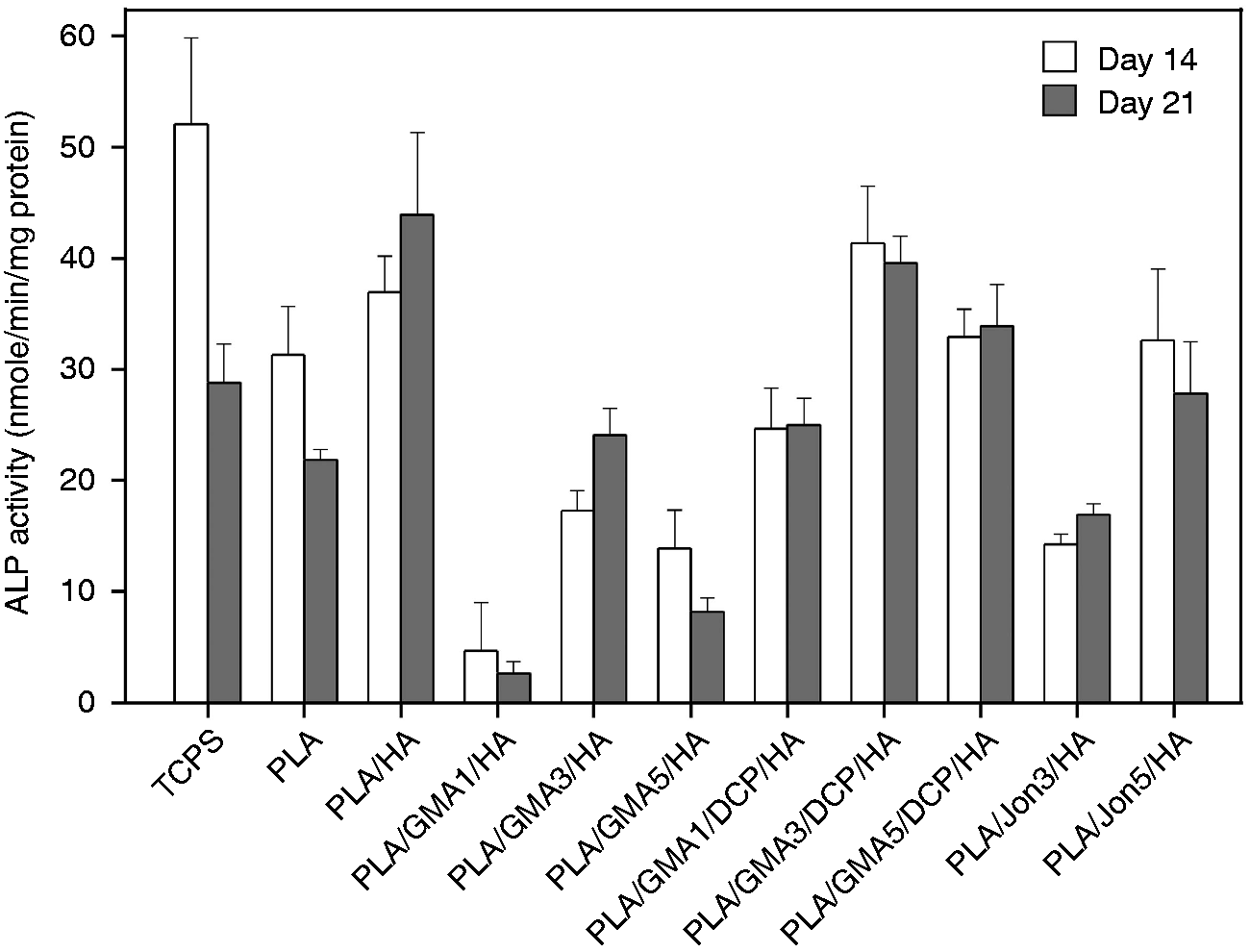
Osteoblast mineralization in MC3T3-E1 cells on different PLA/HA composite and TCPS surfaces after 21 days of culturing.
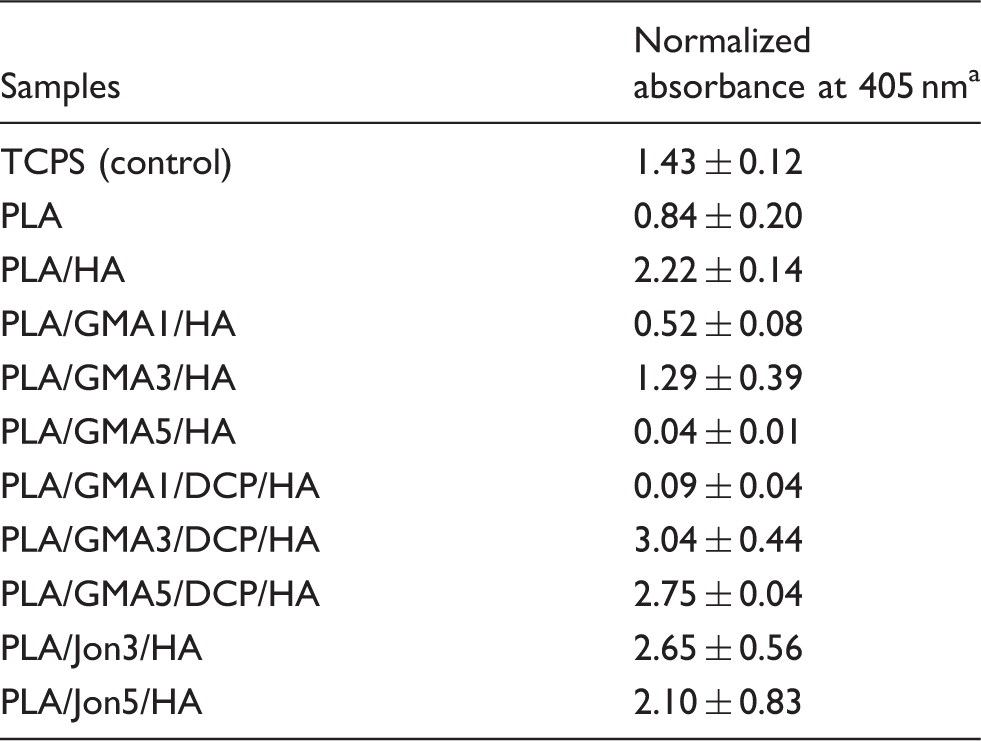
aDue to quantitative differences of surface areas between tested samples (0.25 cm2) and the control (0.32 cm2), the measured absorbance was normalized by surface area.
Conclusions
This work has demonstrated that GMA, a reactive compatibilizer, or Joncryl ADR® containing nine glycidyl methacrylate functions as a chain extension/branching agent affects the flexural strength and strain at break of the 70/30 poly(lactic acid)/hydroxyapatite. With 3 wt.% of GMA and DCP, the flexural strength and strain at break of the composite are obviously increased compared to the composite without GMA or without GMA/DCP. Result from NMR and FTIR insists the occurrence of reaction between glycidyl group of GMA and hydroxyl group of HA which leads to better compatibility and results in higher strength and strain at break. The best result was obtained when Joncryl ADR® contains several glycidyl functional groups. The composite comprising 5 wt.% Joncryl ADR® shows comparable strength to that of PLA but better strain at break. The morphology of the composite with high flexural strength and strain at break shows an agglomeration with long fibrils of PLA. The significant morphology change could be seen in composite with Joncryl where the uniform of HA particles oriented in PLA matrix. The modification of moderate molecular weight through polymer grafting could be the advantage of this composite to be simply fabricated by injection molding technique. Additionally, these PLA/HA composites hold promised as cell-supporting scaffold materials to facilitate cellular attachment, proliferation, differentiation and mineralization. Suitable percentages of the added epoxy functional compatibilizers need to be taken in consideration. Therefore, this PLA/HA composite has a potential to be used as internal bone fixation devices such as screws, pins and plates for implant application.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was performed with the support of the National Metal and Materials Technology Center, the Ministry of Science and Technology of Thailand, Project No. P-1200966.