
Abstract
Encapsulation of therapeutic molecules into nanocarrier is an extensively explored strategy to treat cancer more effectively. In this study, pH-responsive targeting dual-agent delivery nanoparticles were prepared, into which hydrophilic doxorubicin hydrochloride (DOX) and hydrophobic curcumin (CUR) were entrapped. Tyrosine (Tyr) was grafted onto poly(aspartic acid) (PASP) to produce PASP-Tyr, the following reaction between hyaluronic acid (HA) and ethylenediamine (EDA) modified PASP-Tyr formed the nanocarrier HA-EDA-PASP-Tyr (HEPT), and the loading capacity was up to 50.9 ± 4.3% for CUR and 26.0 ± 1.9% for DOX. The spherical HEPT with the mean particle size of 142.9 ± 11.4 nm expanded and deformed into petaloid pattern with an increased size of about 2 µm when triggered by the acidic microenvironment. In vitro anticancer activity evaluation revealed that the co-loaded (DOX+CUR)@HEPT nanoparticles presented higher cytotoxicity against HCT-116 cells compared with that of the free combination of (DOX+CUR). Confocal laser scanning microscopy observation indicated that HEPT carrier promoted cellular uptake of drugs by means of active targeting capacity of HA ligand. With high loading capacity and tailored carrier structure, the nanoparticles formulations may offer a new strategy for cancer treatment.
Introduction
Chemotherapy is one of the three major means to treat cancer, although always accompanied by severe adverse effects.1,2 Most anticancer drugs are associated with a number of drawbacks, including lack of targeting capability, multi-drug resistance (MDR), and poor water solubility. 3 Drug carrier technology is believed as an effective way to circumvent these hurdles and is widely concerned by researchers. Over the past decade, nanoparticle-based drug delivery platforms have been developed and remarkable progress has been made to maximize the efficacy of nanocarrier.3,4 Polymer micelles system, which generally consists of amphipathic molecules, is one of the most promising drug carrier platforms with solubilized hydrophobic drugs, the controllable biological activity of drug, and the potential to overcome biological barriers effectively. In these cases, drugs enter into the cell interior favorably and exert their therapeutic effect better, and preclinical and clinical trials have been ongoing.5,6
Polyamino acid has good application prospects in drug delivery systems because of its excellent biocompatibility and biodegradability. Poly(aspartic acid) (PASP) is a hydrophilic polymer formed by condensing the amino group and carboxyl group of aspartic acid (ASP) monomer, and unreacted carboxyl groups in PASP make it easy to be further modified. 7 Tyrosine (Tyr), another kind of amino acid, can form proteins like ASP, but has a poor affinity with the aqueous phase because of its hydrophobicity. The graft of Tyr onto PASP structure produced amphiphilic polypeptide chains in which PASP served as hydrophilic segment and Tyr acted as a hydrophobic part. When the amphiphilic polypeptide chains self-assembled in water to form micelles, hydrophobic drugs are encapsulated into hydrophobic core, while hydrophilic drugs are bound to the hydrophilic shell, creating a dual-drug delivery system.5,8
Hydrophilic doxorubicin hydrochloride (DOX) is a periodic nonspecific drug with excellent antitumor effects for a wide range of malignant tumors by inhibiting the synthesis of RNA and DNA.9,10 But its clinical application has been limited in part owing to cardiac toxicity and MDR. 11 Hydrophobic curcumin (CUR), an acid polyphenolic substance extracted from turmeric, has a potential myocardial protective effect and displays significant anticancer effects,12–14 inhibiting cancer cell proliferation by blocking multiple cell signaling and genetic pathways. 15 Unfortunately, CUR has a limited application range compared with DOX on account of its poor solubility and rapid metabolism which deteriorates its bioavailability. Nevertheless, pleiotropic CUR, as a reverse agent and/or chemosensitizer, plays key roles in decreasing MDR by downregulating the intracellular level of three major ATP-binding cassette drug transporters, and its combination with DOX presents better treatment efficacy when administered with a delivery system. 16
Efficient delivery of chemotherapy drugs is related to the tumor microenvironment as well as the physical and chemical properties of nanoparticles themselves. 17 To take advantage of pathophysiological conditions, great efforts have been made to improve the treatment efficiency by passive and active targeting, stimuli response, carrier particle properties (size, shape, surface chemistry) modulating as well as combination therapy, and so on. The suitable particle size of 30–200 nm is believed prerequisite for the enhanced permeability and retention (EPR) effect-based passive targeting, 1 and at the same time, a large number of researches have been carried out on cancer cell specific receptors, among which the idea that CD44 receptors overexpress on the surface of cancer cells has been widely recognized. 18 SCC-7, HCT-116 and B16F10 cells all have overexpressed CD44 receptors, while hyaluronic acid (HA) is a recognized target ligand that can specifically bind to them.19–21 Besides, stimuli-sensitive nanoparticles formulations based on the specific tumor microenvironment such as the spatial variations in pH value, relative hypoxia, and local temperature increase have been rationally designed and their drugs delivery efficiency has been evaluated. Given the complexity of delivery and therapeutic administrations, nanoplatforms with multiple functions have been developed, such as peptide-based prodrug combining active targeting and redox-response, 19 polypeptide-based combination micelles integrating pH-response and active targeting, 22 triblock copolymer carriers possessing redox- and pH-sensitivity, 23 amphiphilic micelles delivering multi-drug by pH- and redox-responsive vehicles, 24 and so on. To overcome biological barriers, it is desirable to design an effective nanoplatform integrated with multiple functionalities: passively and selectively targets the cancerous tissues, responds to the tumor microenvironment, and co-administers multi-drug delivery aiming to acquire better synergistic effect.
In this study, a polyamino acid-based co-delivery system was presented, which encapsulated both hydrophobic CUR and hydrophilic DOX, integrated pH-response, passive and active targeting into one nanoplatform as shown in Figure 1. Tyr was grafted onto the side chain of PASP by amidation reaction and served as a hydrophobic part in the carrier, the influence of graft rates on the loading capacity for CUR was investigated to help understand the loading mechanism of the hydrophobic drug. The reaction between the carboxyl group of HA and the amino group in ethylenediamine (EDA) bound HA ligand onto PASP-Tyr (Figure 2). The collapsed carrier structure in acidic microenvironment released two drugs with optimal weight ratio, which was beneficial to the promotion of anticancer performance synergistically.
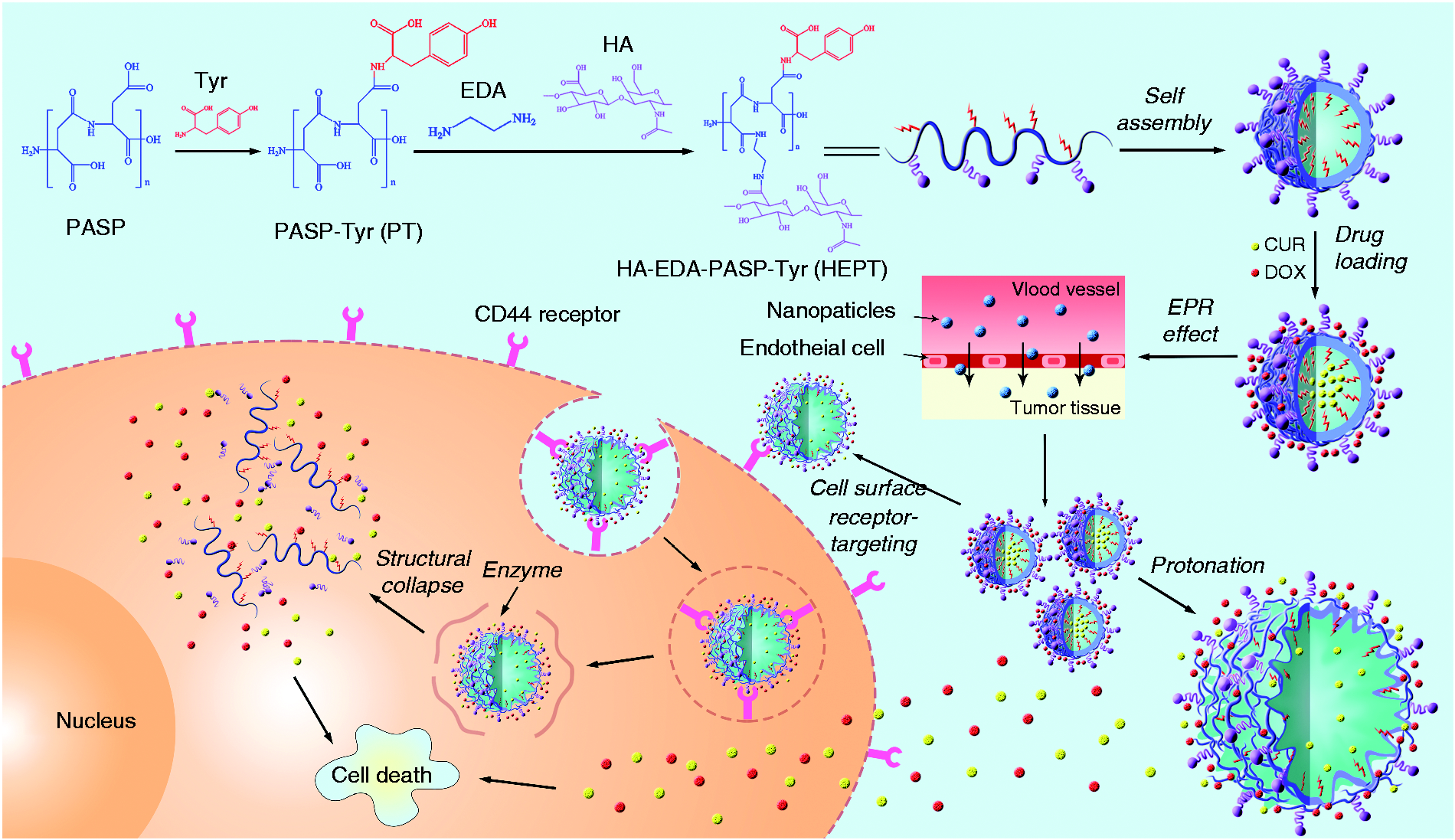
Schematic representation of preparation process and action principle of targeting and pHresponsive self-assembled multi-drug carrier.
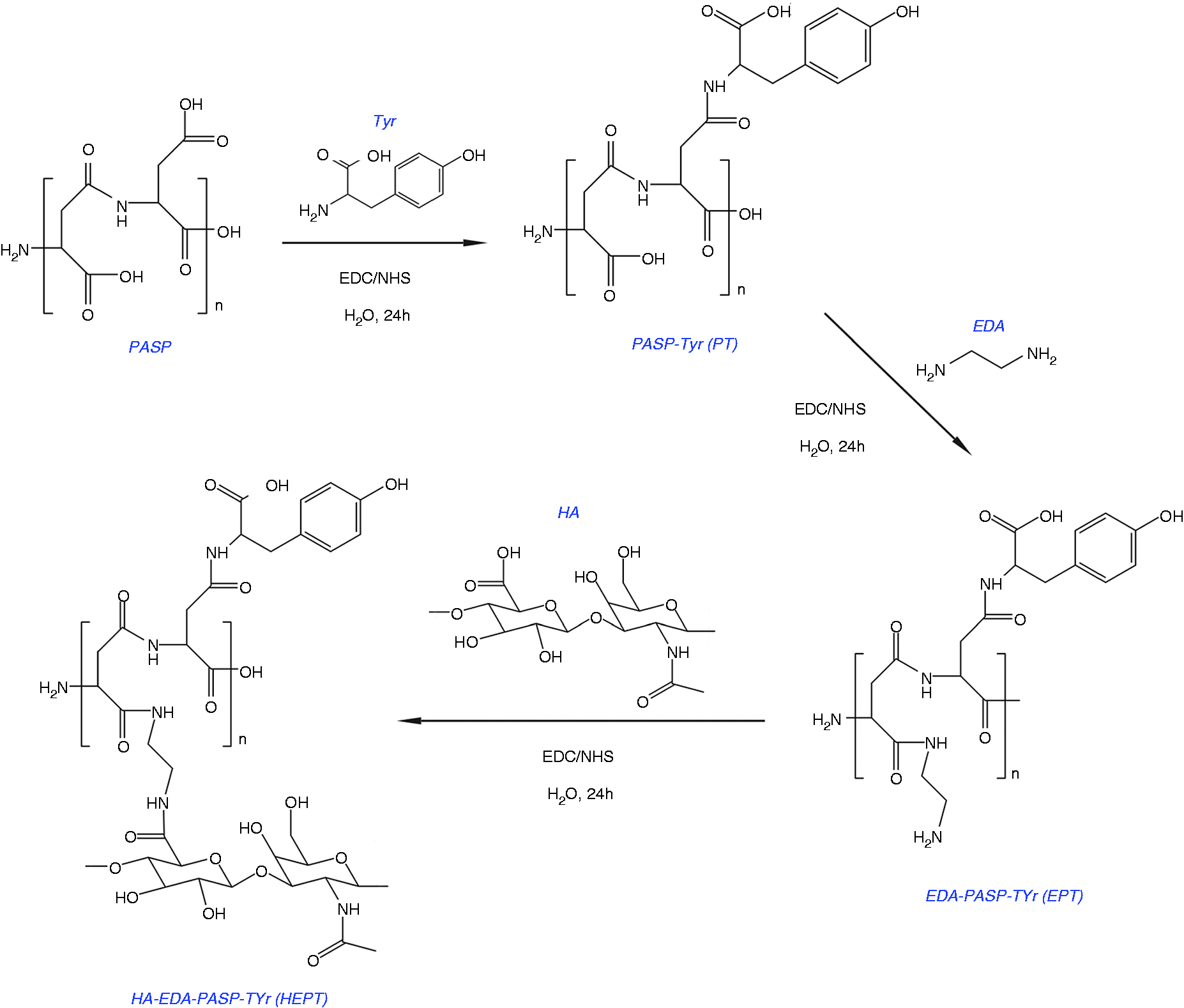
The synthetic route of PT and HEPT.
Materials and methods
Materials
PASP was purchased from Shandong West Asia chemical industry co., LTD.), Tyr, N-(3-dimethylaminopropyl)-N-ethylcarbodiimide hydrochloride (EDC·HCl), N-hydroxysuccinimide (NHS), DOX, paraformaldehyde, 4′6-diamidino-2-phenylindole (DAPI), and Trypsin were supplied by Aladdin (Shanghai, China), EDA, HA and CUR were obtained from Tianjin kemio chemical reagent co., LTD.), human colon cancer cell line HCT-116 was provided by Chinese Academy of Sciences, medium (McCoy’s 5 A) was obtained from Vicente biotechnology (China) co., LTD. and DMSO was supplied by Sinopharm group chemical reagent co., LTD.
Methods
Proton nuclear magnetic resonance (1H NMR) was determined at 25 °C using a Bruker AVANCE III 400 MHz NMR spectrometer with D2O as a solvent. Fourier transform infrared spectroscopy (FT-IR) was obtained on a TENSOR 27 FT-IR spectrometer (Bruker, Germany). The drug loading capacity was determined by ultraviolet-visible spectrophotometer (UV–Vis spectrophotometer, UV-1800, Shimazu enterprise management co., LTD.). The average particle size and zeta potential were detected by using dynamic light scattering (DLS, S3500, Microtrac Inc, USA). The morphologies of the carrier were observed by high resolution transmission electron microscope (HR-TEM, JEM2010, JEOL, Japan) and scanning electron microscopy (SEM, LYRA3XMH, TESCAN, Czech). Automatic surface tensiometer JK99B (Shanghai zhongchen digital technology equipment co., LTD.) was employed to determine the critical micelle concentration (CMC). Optical density value was obtained on a microplate reader (Nivo, PerkinElmer). Cellular uptake was investigated by confocal laser scanning microscopy (CLSM) (C2 Plus, Nikon, Japan).
Synthesis of PASP-Tyr
0.36 g of Tyr (2 mmol) was dispersed in 10 mL of water, followed by sonication for 30 min using a probe-type ultrasonicator. 0.8 g of PASP (0.2 mmol) was dissolved in 20 mL of water followed by the addition of 0.192 g of EDC·HCl (1 mmol) and 0.115 g of NHS (1 mmol) for activation. To this mixture, the as-prepared Tyr water dispersion was added into, and the reaction solution was stirred at room temperature for 24 h. After that, the solution was filtered to remove unreacted Tyr, and dialyzed against deionized water using 2000 Da dialysis bag for 3 days to ensure the complete removal of EDC·HCl and NHS. The PASP-Tyr (PT) powder was obtained after drying and grinding. Changing the weight ratio of PASP, Tyr, EDC·HCl and NHS produced PT with different grafting rates.
Synthesis of HA-EDA-PASP-Tyr
80 mg of as-prepared PT powder (0.02 mmol) was dissolved in water, and 19.2 mg of EDC·HCl (0.1 mmol) and 11.5 mg of NHS (0.1 mmol) were added into. After activation at room temperature for 40 min, a drop of EDA was added into, and the reaction between PT and EDA was carried out for 24 h at room temperature, followed by dialyzing against deionized water for 3 days. EDA-PASP-Tyr (EPT) powder was collected after drying and grinding consecutively. Afterward, 100 mg (0.25 mmol) of HA, 48 mg of EDC·HCl (0.25 mmol), and 29 mg of NHS (0.25 mmol) were dissolved in water under stirring at room temperature for 40 min. Thereafter, 100 mg of EPT powder (0.025 mmol) was added to the above solution, and reaction was carried out at room temperature for 24 h. After the reaction, the mixture was purified by dialyzing (MWCO: 2000 Da) against deionized water for 3 days to remove unreacted HA, EDC, and NHS. In the end, HA-EDA-PASP-Tyr (HEPT) powder, the drug carrier, was obtained by drying and grinding consecutively.
Determination of CMC
There are two main strategies to prepare self-assembled micelle particles, one of which is the ultrasonic dispersion method (direct preparation method), and another one is the dialysis method. 25 This section described the direct preparation method without drug loading process. The as-prepared carrier (PT or HEPT) was dissolved in water and subjected to ultrasonic treatment at room temperature for 10 min. In this way, PT or HEPT formed self-assembled micelle particles in the aqueous surrounding. To determine CMC, PT, or HEPT solution with different concentration gradients was prepared, and the surface tension was measured and plotted against the logarithm of concentration, the concentration corresponding to trend inflection point was CMC.
Loading of CUR
The loading of CUR was carried out by dialysis method. 25 12 mg of CUR was dissolved in 20 mL of anhydrous ethanol at 0.6 mg/mL, and 10 mg of PT or HEPT was added into and mixed by ultrasonic stirring for 30 min followed by an additional 24 h stirring under an ice bath in the dark. Thereafter the well-mixed dispersion was dialyzed (MWCO: 2000 Da) against deionized water for 3 days, and anhydrous ethanol inside dialysis bag was gradually replaced by water. As a consequence, PT or HEPT formed micelles with the hydrophobic core and hydrophilic shell region in the aqueous surrounding. The hydrophobic core facilitated the solubilization of poorly soluble CUR and the hydrophilic outer surface provided sites for soluble molecules. Unloaded CUR was removed by dialyzing, and the drug-loaded carrier powder CUR@PT or CUR@HEPT was obtained by freeze-drying.
Loading of DOX
DOX was loaded by the physical blending method. Briefly, DOX was dissolved in deionized water at 0.6 mg/mL, and CUR@HEPT (10 mg) was added into under ultrasound dispersion for 30 min followed by 24 h stirring in the dark. The unloaded DOX was dissolved in supernatant so that it can be removed by centrifugation, and the drug co-delivery vehicle, denoted as (CUR+DOX)@HEPT, was collected by freeze-drying the precipitate. Thus, polyamino acid-based drug co-delivery system encapsulated both hydrophobic CUR and hydrophilic DOX simultaneously was obtained. As DOX has photodegradation property, special attention during handling and storage in solution form is needed, and the procedure should be kept away from light.
Drug loading capacity and in vitro drug release curve
The drug concentration was measured at 425 nm for CUR and 480 nm for DOX by UV–Vis spectrophotometer, then the drug encapsulation efficiency, and the drug loading capacity was calculated according to the following equation:
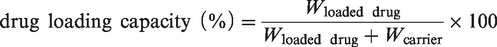

The drug release was performed in different pH solutions in the dark. Phosphate buffered saline (PBS) of pH 7.4 and 5.4 was used as the drug-release media to simulate normal blood/tissue and tumor environments. The (CUR+DOX)@HEPT nanoparticles were dispersed in 5 mL of release media, after that the suspensions were placed into pretreated dialysis bags (MWCO = 2000 Da). The dialysis bags were placed into brown bottles to which 100 mL of PBS was added. The bottles were shaken at 100 rpm at 37 °C away from light. PBS was replaced with a fresh buffer solution at predetermined time points. The released drugs were examined by UV–Vis spectrophotometer.
The in vitro drug release rate was calculated according to the following equation and drug release curves were constructed:
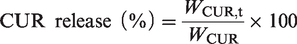
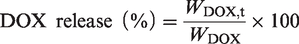
In vitro cytotoxicity
Cytotoxicity studies were performed with both free drugs and the (CUR+DOX)@HEPT nanoparticles. Cell growth medium was composed of McCoy's 5 A, 10% FBS, and 1% P/S. HCT-116 cells were used for in vitro proliferation. Briefly, 2000 cells per well were plated in 96-well plates and incubated overnight in an incubator containing 5% CO2 at 37 °C. Then, pure HEPT, the combination of CUR and DOX (named as (CUR+DOX)) and co-delivery vehicle (CUR+DOX) @HEPT were added to wells and incubated in an incubator containing 5% CO2 at 37 °C for 72 hours in the dark. After that, CCK8 (10 µL per well) was added to the cell well plate and incubated in an incubator containing 5% CO2 at 37 °C for 2 ∼ 4 hours. The absorbance at 450 nm wavelength was monitored at a microplate reader and the cell viability was determined by comparison with untreated cells and calculated according to the following formula:
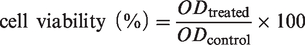
Cellular uptake
Cellular uptake study was investigated by CLSM. Firstly, HCT-116 cells were seeded in 24-well plates (1 × 104 cells per well) and incubated in 2 mL of culture medium for 24 h at 37 °C in a humidified atmosphere containing 5% CO2. Subsequently, the (CUR+DOX)@HEPT or free drug combination (CUR+DOX) were added, and the cells were allowed to incubate for another 2 h. After removing the medium and washing with PBS solution thrice, the cells were fixed with 4% paraformaldehyde for 10 min. Afterward, the cells were counterstained cell nucleus by DAPI for 10 min and viewed under CLSM.
Enzymatic degradation of HEPT
Trypsin was employed as the degradation enzyme. Trypsin was dissolved in PBS (pH7.4) and the concentration was set as 250, 500, 750, 1000, and 1250 U/mL. The HEPT was added into the enzyme solution, and the mixture was shaken at 80 rpm at 37 °C, from which the sample was withdrawn at a predetermined time interval. The remained HEPT ratio was examined by UV-Vis spectrophotometer at 210 nm.
Statistical analysis
Data were presented with mean ± standard deviation (S.D.) of the mean. The statistical significance was determined by t-test analysis of variance. A value of P < 0.05 was considered statistically significant.
Results and discussion
Synthesis of PASP-Tyr and HA-EDA-PASP-Tyr
The synthesis of PT and HEPT was verified by 1H NMR) and FT-IR. According to 1H NMR (Figure 3), the chemical shift at 2.4 ∼ 2.7 ppm (1) was assigned to the methylene proton of PASP. The characteristic signal peak at 3.0 ppm (2) was attributed to the methylene proton from Tyr, which proved that Tyr was grated onto PASP. The peak located at 1.9 ppm (3) was ascribed to methyl proton from HA, 26 and proton signal peaks at 3.27 (4), 3.41 (5), 3.63 (6), and 3.72 ppm (7) were attributed to methylene proton from HA, respectively. These spectra results demonstrated the successful synthesis of HEPT. The formation of PT and HEPT was further supported by FT-IR spectra (Figure 4). Compared with PASP, the peak for PT at 900 ∼ 934 cm−1 was attributed to the out-of-plane bending vibration peak of C-H from benzene ring in Tyr. Compared with PT, the peak for HEPT at 1041 cm−1 and 1080 cm−1 was attributed to the stretching vibration peak of C-OH and carbonyl in HA, respectively. Besides, the characteristic stretching vibration peak of C-O-C in HA located at 1149 cm−1 was observed. Both 1H NMR and FT-IR outcomes suggested the synthesis of HEPT carrier.
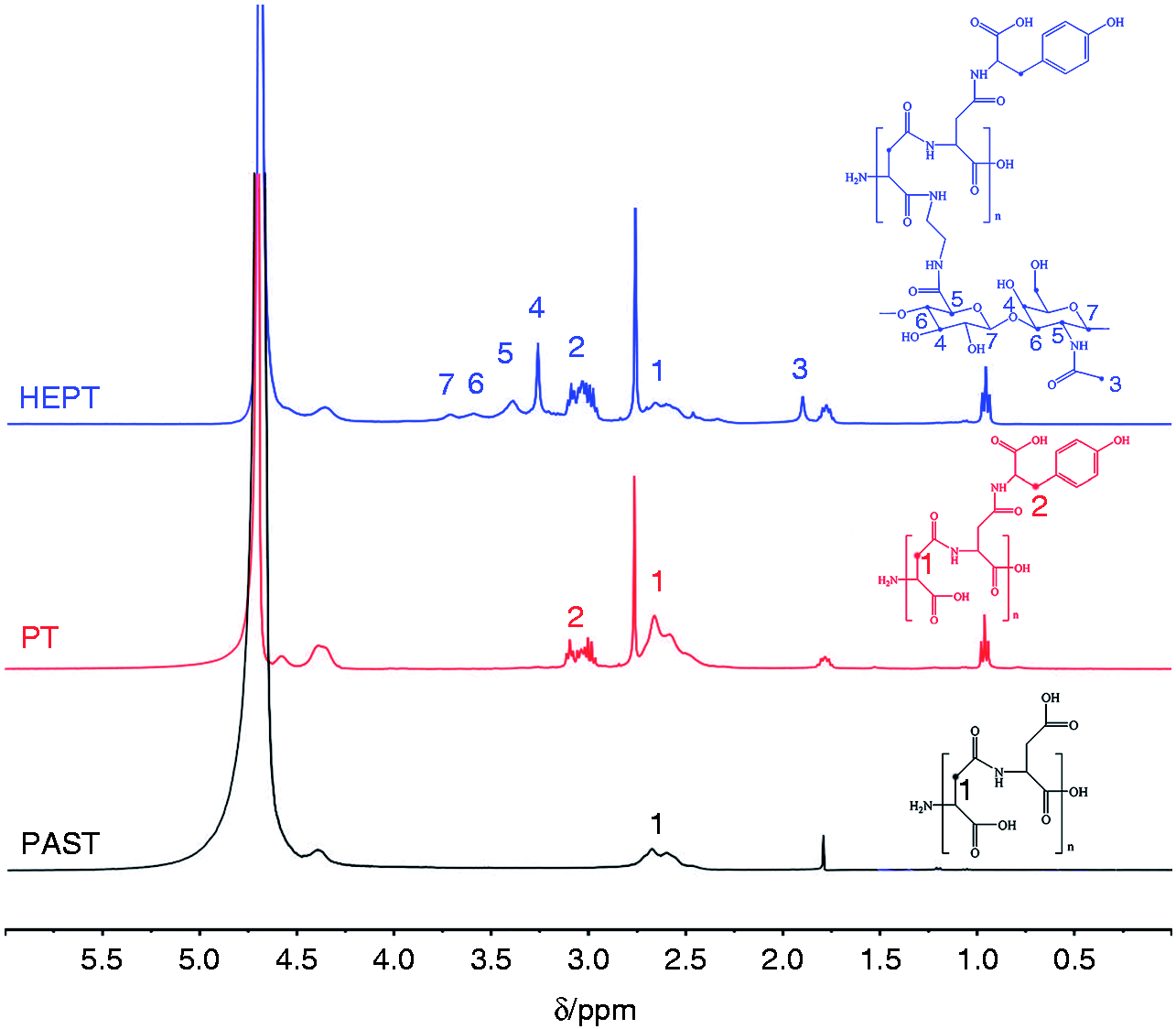
1H NMR spectra of PASP, PT and HEPT.
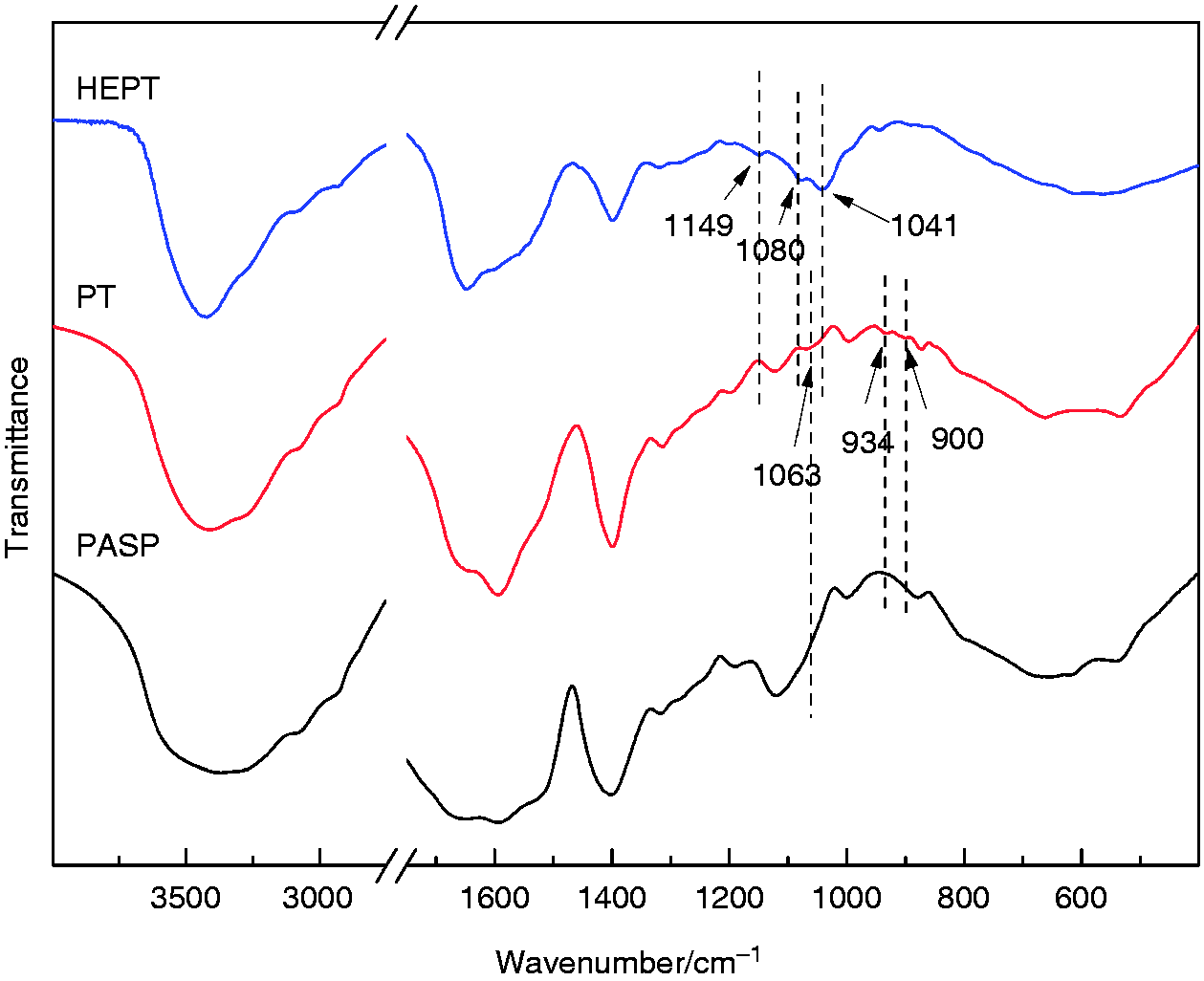
FT-IR spectra of PASP, PT and HEPT.
Characterization of HEPT self-assembled micelle nanoparticles
The CMC of HEPT was tested by surface tension method, and the surface tension was used to make a scatter diagram against the concentration logarithm (Figure 5). The inflexion point was obtained by fitting, the corresponding lgC was −1.314, indicating the CMC was calculated to be 0.049 mg/mL. In this study, the concentration of HEPT was always much higher than CMC to ensure stable micelles formation. 25
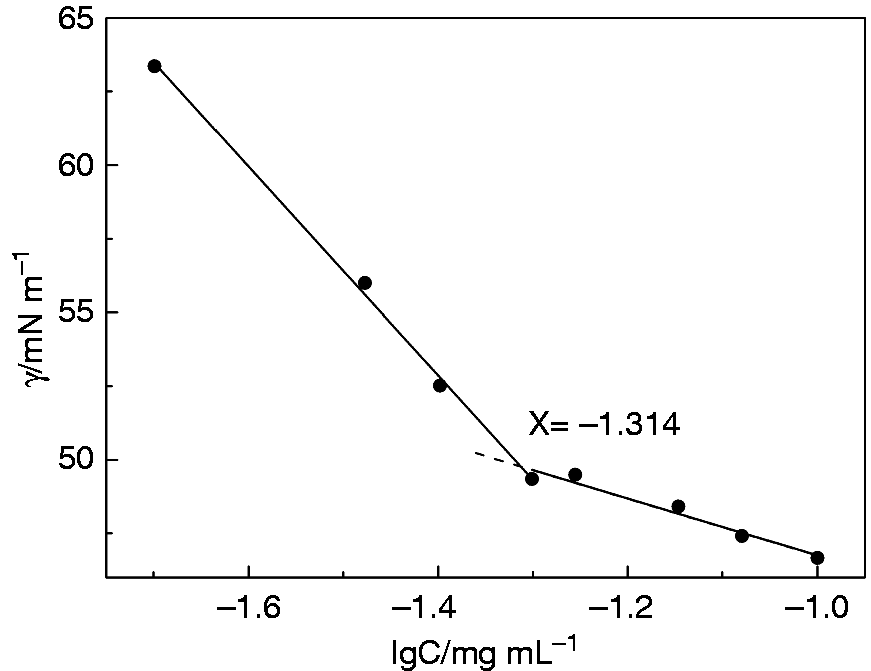
The image of relationship between surface tension and logarithmic concentration of HEPT. Data are presented as mean ± SD (n = 3).
The morphology of micelle nanoparticles was observed by HR-TEM and SEM, which displayed a nearly spherical shape and uniform size distribution (Figure 6). The average particle size was about 142.9 ± 11.4 nm with a polydispersity index intensity (PDI) of 0.257 (Figure 7(a)), which was believed to be beneficial to take advantage of EPR effect. 1 When pH was set at 5.4, the wide particle size distribution suggested that the carriers underwent varying degrees of expansion in acidic conditions (Figure 7(b)). The expansion was associated with protonation in acidic conditions, which reduced intermolecular force, leading to expanded and loose structure, triggering the release of drugs wrapped in the micelles. Taking advantage of the pathophysiological conditions such as the EPR effect and pH variations, the HEPT carrier, with suitable size and pH-sensitive characteristics, facilitated the drug release in the acidic extracellular environment of solid tumor.

(a) TEM and (b) SEM images of (CUR+DOX)@HEPT nanoparticles at pH 7.4.
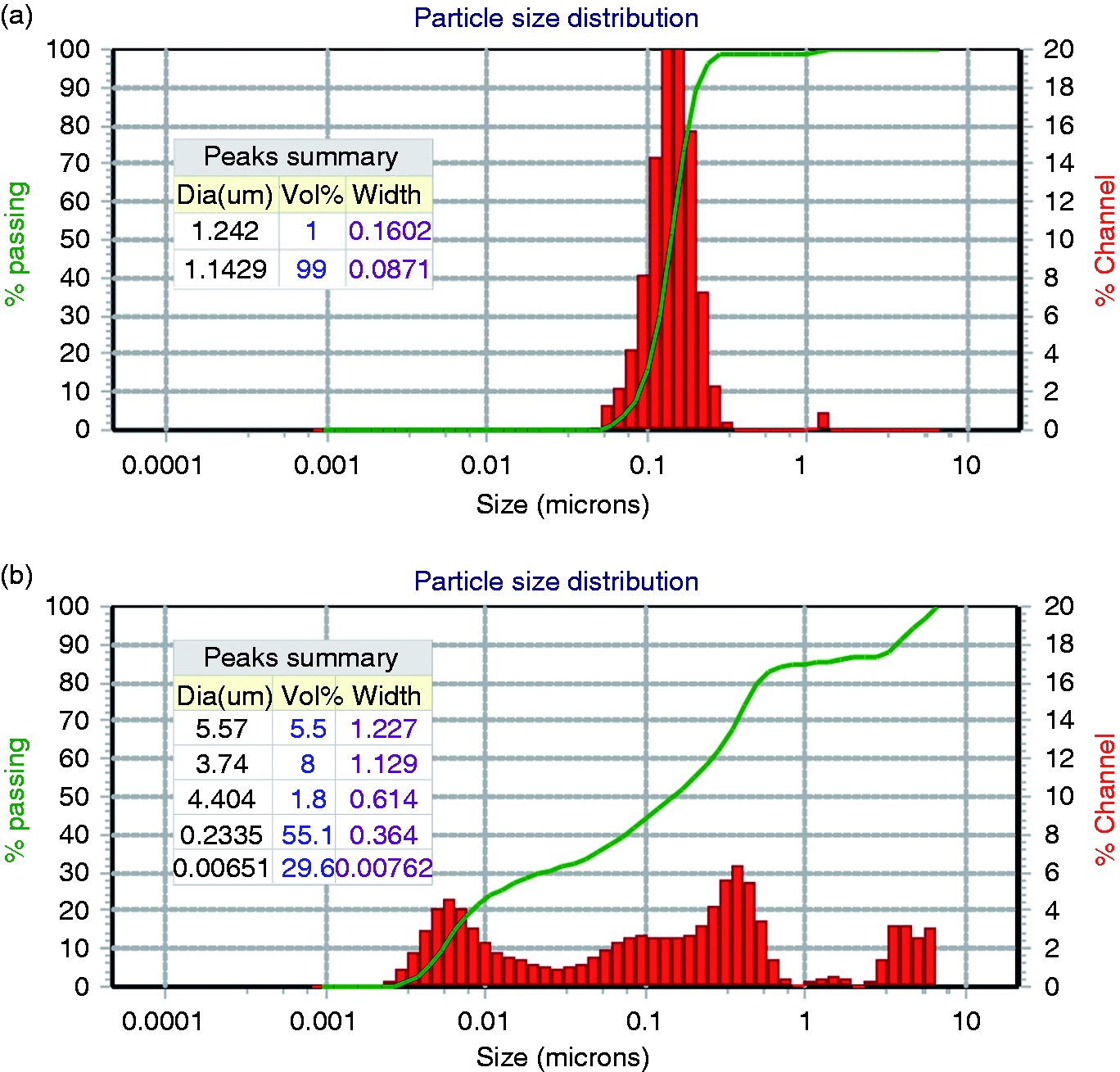
The particle size distribution of HEPT at (a) pH 7.4 and (b) pH 5.4.
Effect of graft rates on drug loading capacity
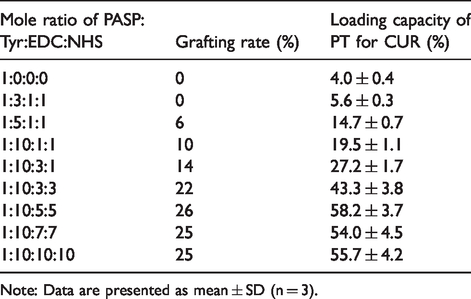
PT was synthesized by reacting PASP and Tyr with EDC·HCl and NHS. Changing the weight ratio produced PT with different grafting rate, which affected the carrier loading capacity, especially for hydrophobic CUR. The Tyr graft rate, tuned by feeding ratio, was determined by the integral ratio calculated according to 1H NMR (Figure 8) by comparing the peak area between the methylene proton (2) of PASP and the methylene proton (1) from Tyr as shown in Table 1.
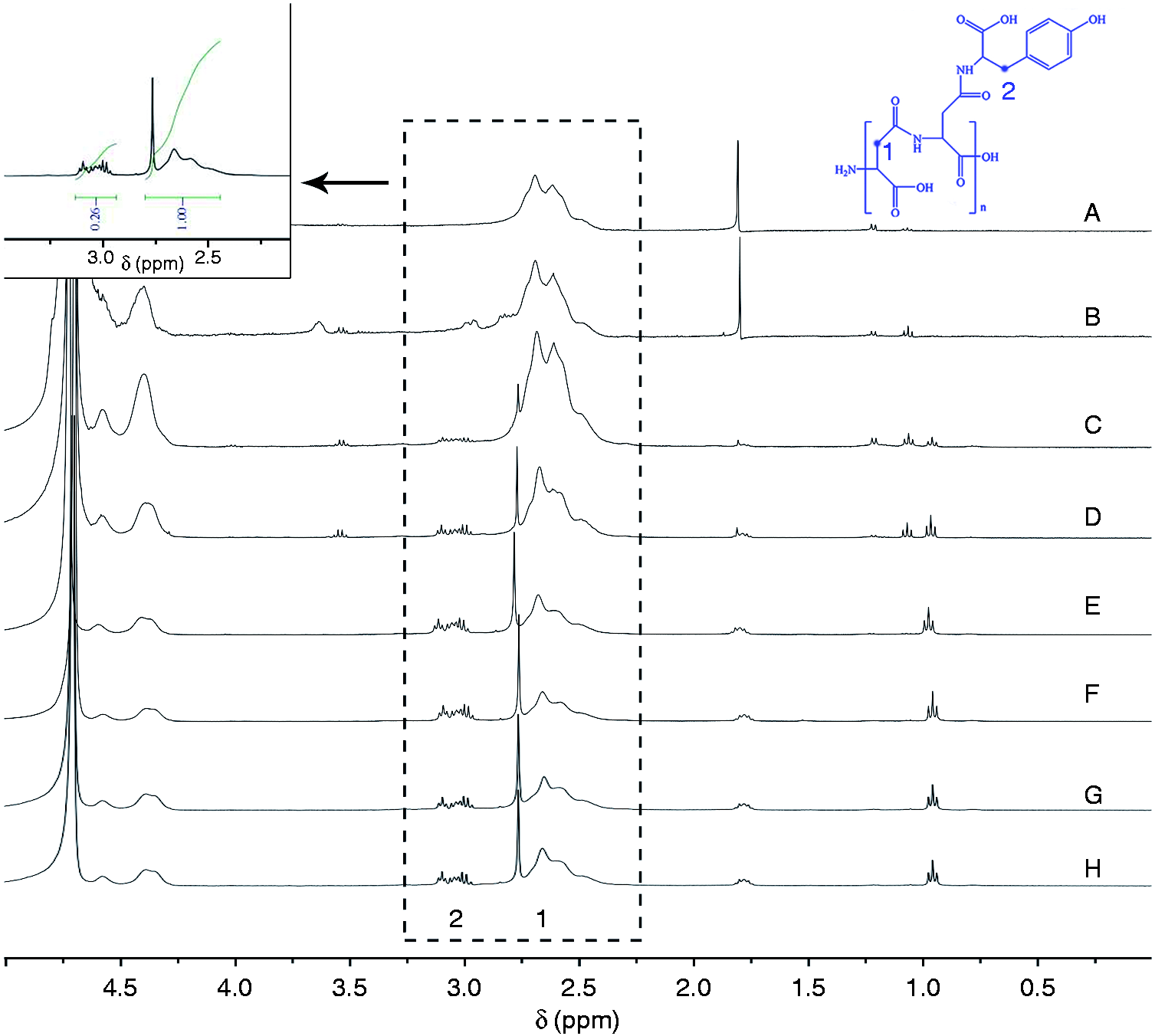
1H NMR spectra of PASP-Tyr at different feeding ratios (PASP:Tyr:EDC:NHS). (A: 1:3:1:1, B: 1:5:1:1, C: 1:10:1:1, D: 1:10:3:1, E: 1:10:3:3, F: 1:10:5:5, G: 1:10:7:7, H: 1:10:10:10).
Effect of feeding ratios on graft rate and drug loading capacity for curcumin.
Note: Data are presented as mean ± SD (n = 3).
With the increase of the amount of Tyr and EDC/NHS, the graft rate increased, which resulted from the reaction principle of the EDC/NHS system. EDC plays the role of activating carboxyl groups, while NHS transforms the activated carboxyl groups into active esters, thus stabilizing the activated carboxyl groups. Therefore, increasing the amount of EDC increased the amount of activated carboxyl group, while increasing the amount of NHS increased the stable activated carboxyl group, leading to elevated graft rate.27,28 Notably, the increased grafting rate resulted in improved drug loading capacity for CUR owing to the special micellar structure. On one hand, the increase of hydrophobic part (Tyr) in PT molecules enhanced the π-π stacking interaction between carrier and CUR. On the other hand, the increase of the hydrophobic part raised the steric hindrance of the hydrophobic core and enlarged the micellar internal space which served as the reservoir for hydrophobic drugs in aqueous solutions. To evaluate the loading capacity, CUR was loaded onto PT firstly by dialysis method, followed by DOX loading. When the mole ratio of PASP:Tyr:EDC:NHS was 1:10:5:5, the drug loading capacity of PT carriers was calculated to be 29.8 ± 2.3% and 48.8 ± 2.8% for CUR and DOX, respectively. Accordingly, the drug encapsulation efficiency of CUR and DOX were 76.5 ± 6.7% and 84.3 ± 5.3%, respectively. The high drug loading capacity for CUR is not only attributed to the self-assembly and encapsulation by the carrier but also the π-π stacking interaction between the benzene rings existing both in Tyr and CUR. After grafting of HA onto PT, the loading capacity for DOX decreased from 48.8 ± 2.8% to 26.0 ± 1.9%, resulting from the fact that HA occupied part of sites in PASP hydrophilic segment. Consequently, the loading capacity in HEPT were 50.9 ± 4.3% for CUR and 26.0 ± 1.9% for DOX, and the drug encapsulation efficiency of CUR and DOX were 77.8 ± 5.5% and 53.7 ± 4.8%, respectively. The corresponding weight ratio of chemosensitizer CUR and anticancer agent DOX was close to 2:1, such a ratio presented remarkable therapeutic efficacy in MDR cells and decreased normal tissue drug toxicity, providing a better synergistic effect.29,30
In vitro drug release
The in vitro release profiles of CUR and DOX from (CUR+DOX)@HEPT nanoparticles at solutions with different pH values were shown in Figure 9. An obvious pH-response effect was observed, and CUR was more sensitive to pH variation (Figure 9(a)). At 24 h, CUR showed slow release at pH 7.4 (26.0 ± 0.7%), but fast release at pH 5.4 (87.0 ± 2.4%) with a 2.35-fold increase. CUR was wrapped inside the micelles by hydrophobic interaction and such a binding force was relatively poor. Once the micellar structure disintegrated in an acidic environment, a burst release occurred. The interaction between DOX and carriers included hydrogen bond, charge adsorption, and so on, which were relatively strong than the case of CUR, so the release was slow even if the carrier structure was collapsed. But DOX still presented the pH-sensitive feature to a certain extent, and fast release at pH 5.4 (81.4 ± 2.2%) was detected compared with that at pH 7.4 (74.2 ± 3.1%), which may mainly arise from the weakened charge interaction between carrier and positively charged DOX in an acidic environment. The weight ratio of released CUR and DOX from (CUR+DOX)@HEPT in acidic condition was calculated and showed in Figure 9(b). The average weight ratio was 2.149:1 at pH 5.4, and this value decreased to 0.763:1 at pH 7.4. This phenomenon revealed that the dual-agent loaded (CUR+DOX)@HEPT system released CUR and DOX in proportion of about 2:1 in an acidic microenvironment, in accord with their weight ratio encapsulated in. The special structure of HEPT carrier and its pH-response property played a key role in the proportional releasing in acidic conditions. The highest weight ratio of 2.602:1 appeared in the first hour, which was probably related to such a fact that the collapse of the carrier in acidic condition released a large amount of CUR from the core of nanoparticles compared with that at pH 7.4, leading to an increased weight ratio of CUR and DOX. The typical morphology of HEPT with different immersion times in acidic condition was observed by HR-TEM (Figure 9(c)), which revealed gradually increased particle size and disintegrated structure, facilitating the progressive rise of CUR concentration. At the same time, the expanded carrier size and loose structure increased contact surface areas of the hydrophilic segment with the outer acidic microenvironment, promoting DOX release. Of course, the several expanded carrier shapes may exist simultaneously, while it was well-founded to suppose that carrier morphology progressively changed over time in acid conditions. The subsequent releasing process for both CUR and DOX was relatively steady and maintained the desired release profile with releasing weight ratio of about 2:1, which was optimal to kill cancer cells synergistically.
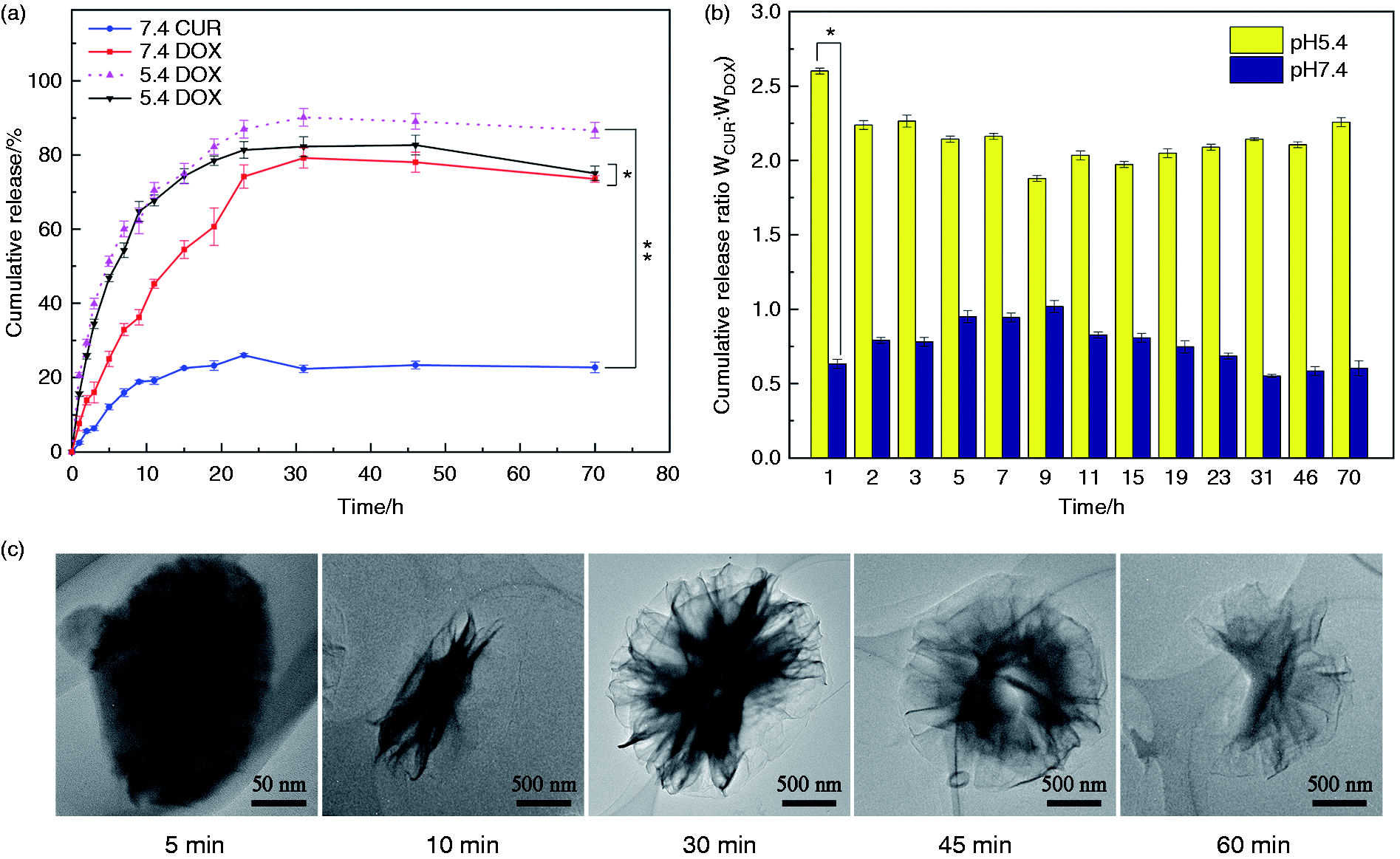
(a) Curve of in vitro cumulative release of CUR and DOX from (CUR+DOX)@HEPT at different pH value over time. (b) Weight ratio of released CUR and DOX from (CUR+DOX)@HEPT at pH5.4 and 7.4. (c) TEM images of (CUR+DOX)@HEPT particles with increasing immersion time in acid environment. Data are presented as mean ± SD (n = 3). *p < 0.05; **p < 0.01.
Zeta potential was tested to investigate nanoparticles stability at different pH (Figure 10). The HEPT nanoparticles were negatively charged and zeta potential was −17.1 ± 1.3 mV at pH 7.4, indicating a relatively stable micelle system. With the decrease of pH, the zeta potential increased, and it reached −3.4 ± 0.5 mV at pH 5.4, implying poor stability because of the protonation of micelle in an acidic environment, in accordance with its pH-sensitive drug release profile.
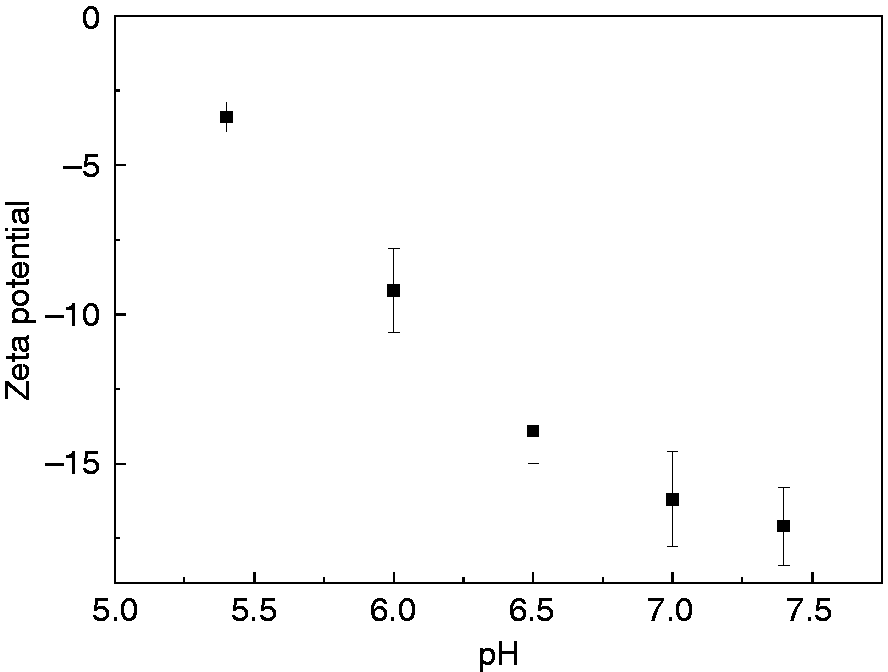
The zeta potential of HEPT at different pH.
In vitro cytotoxicity
In vitro cytotoxicity of the (CUR+DOX)@HEPT nanoparticles against HCT-116 cells was evaluated and shown in Figure 11. Blank HEPT caused cell death scarcely, which implied that cell cytotoxicity of (CUR+DOX)@HEPT was resulted from loaded CUR and DOX components rather than HEPT carrier itself. Free drug combination (CUR+DOX) displayed cytotoxicity as CUR and DOX themselves possessed the antitumor ability, and the cell viability decreased with the increase of drug concentration. The co-encapsulated (CUR+DOX)@HEPT showed higher cell cytotoxicity than that of free drug combination, which was possibly relevant to the action mechanism of (CUR+DOX) @HEPT nanoparticles. Firstly, HA targeting ligand on HEPT carrier could bind to the overexpressed CD44 receptor on HCT-116 cells surface, increase affinity and promote phagocytosis, leading to drug concentration at the cell surface higher than that of free (CUR+DOX), and so higher intracellular drug concentration. Secondly, the encapsulation of hydrophobic CUR into the core of the carrier overcame its poor water solubility, making it reach the same site as DOX at the same time. Finally, the obvious pH-responsive HEPT carrier promoted the rapid localized release of CUR in the acidic microenvironment, leading to a relatively constant weight ratio of CUR and DOX (approximately 2:1), which was believed an optimum value to obtain better synergistic effect. 30
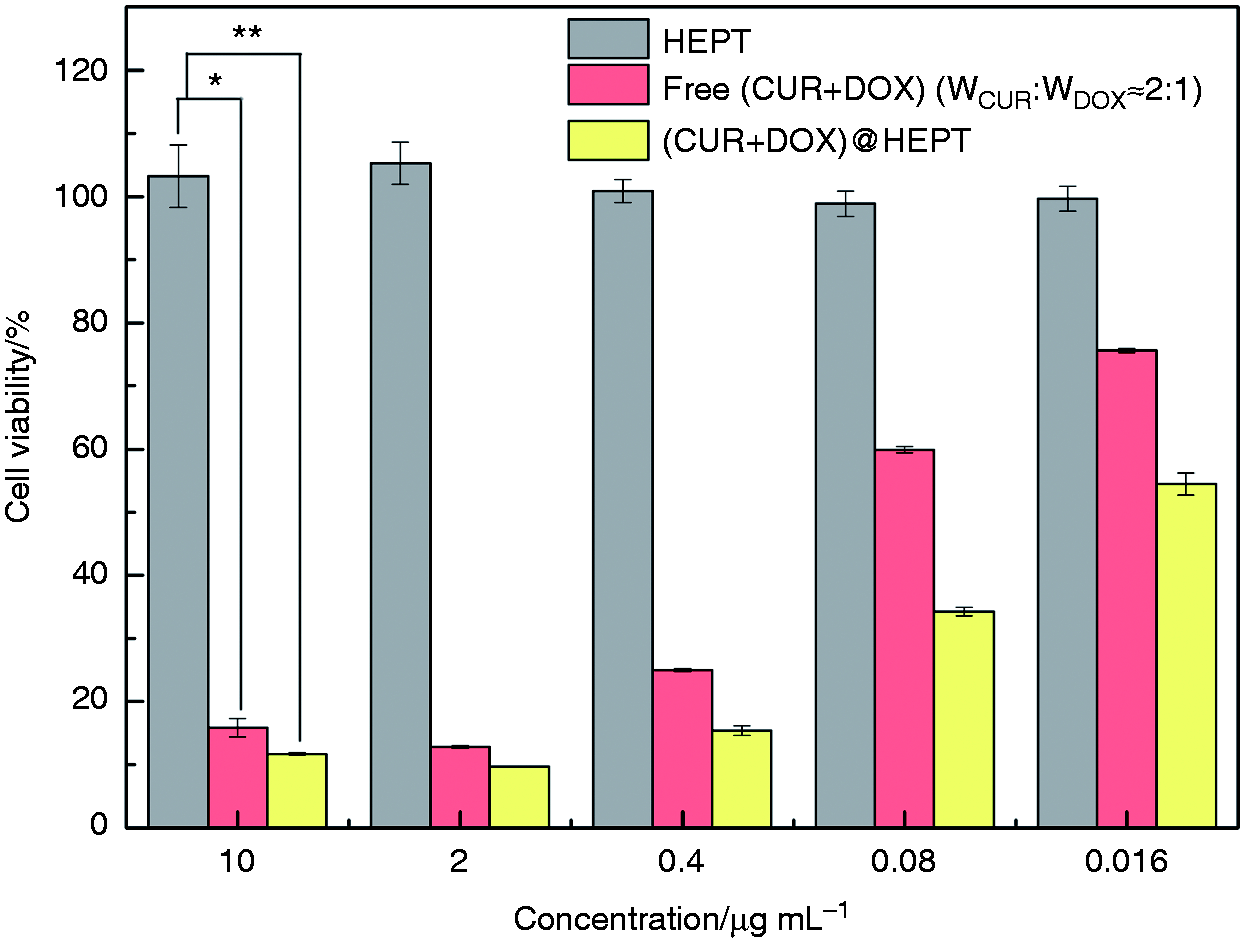
Cell viability of HEPT, free combination of (CUR+DOX) and (CUR+DOX)@HEPT in HCT-116 cells. For samples (CUR+DOX) and (CUR+DOX)@HEPT, the concentration referred to drugs concentration; for HEPT, it referred to the concentration of pure HEPT carrier. Data are presented as mean ± SD (n = 3). *p < 0.05; **p < 0.01.
Cellular uptake
The cellular uptake of (CUR+DOX)@HEPT and free combination of (CUR+DOX) in HCT-116 cells was evaluated by CLSM (Figure 12). Intense green and red fluorescence distribution was found in HCT-116 cells treated with (CUR+DOX)@HEPT, indicating that the CUR and DOX released from nanoparticles were delivered into the nuclei. In comparison, the fluorescence in cells treated with free combination of (CUR+DOX) was weak, and only a relatively small amount of drugs entered into nuclei, leaving some drugs outside. The HEPT carrier promoted cellular uptake of drugs by means of HA targeting ligands.
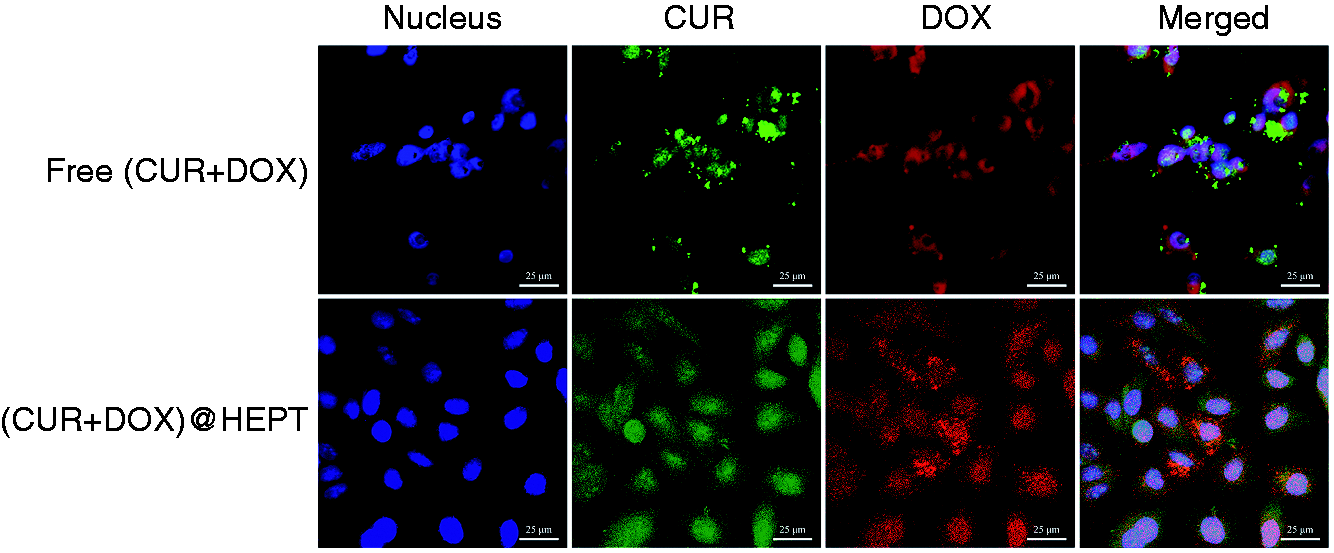
CLSM images of HCT-116 cells incubated with Free (CUR+DOX) and (CUR+DOX)@HEPT nanoparticles for 2 h, respectively. The nuclei of the cells were stained with DAPI (blue).
In vitro degradation
In vitro biodegradability was characterized by an enzymatic degradation experiment. Trypsin could promote the degradation of HEPT carrier as shown in Figure 13. A rapid degradation rate was observed during the first 7 hours, after that, the degradation rate began to level off. The higher enzyme concentration led to fast degradation rate, and the remained ratio was about 46.5 ± 2.1% at trypsin concentration of 1250 U/mL at 24 h.
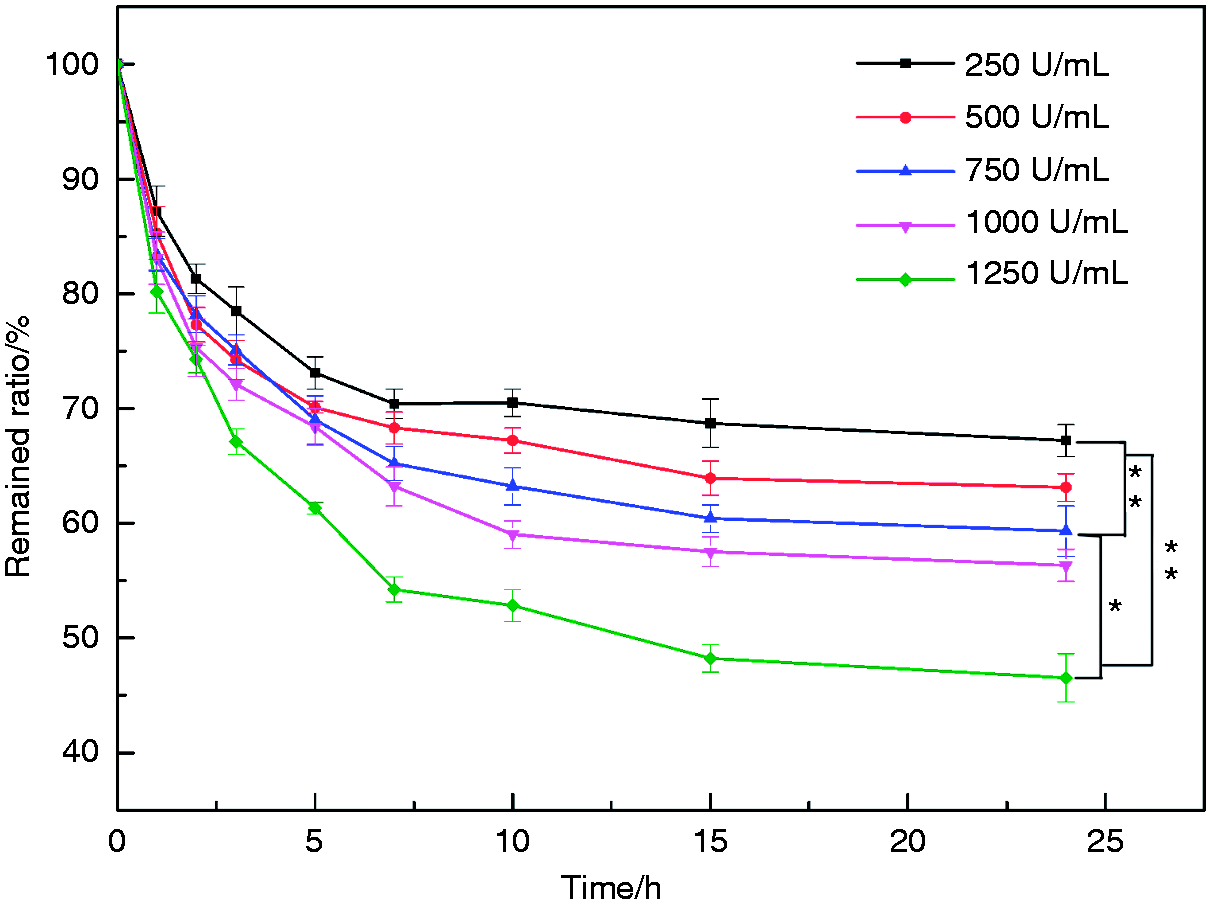
The remained ratio of HEPT over time during enzyme degradation. Data are presented as mean ± SD (n = 3). *p < 0.05; **p < 0.01.
Conclusion
A pH-responsive multifunctional polyamino acid-based micellar drug carrier HEPT encapsulated with CUR and DOX was prepared, the loading capacity was up to 50.9 ± 4.3% for CUR and 26.0 ± 1.9% for DOX. Tyr was grafted onto PASP to form amphiphilic polypeptide chains, and the effect of graft rate on drug loading capacity was discussed aiming to understand the loading mechanism of hydrophobic drugs. The suitable particle size endowed vehicles with passive targeting capacity, and the instinct pH-sensitive HEPT carrier expanded and disintegrated progressively when triggered by the acidic microenvironment, facilitating drugs release with relative steady weight ratio. The HA ligand promoted cellular uptake of drugs released from (DOX+CUR)@HEPT nanoparticles by active targeting capacity. The cytotoxicity test verified that the pH-responsive targeting co-delivery nanoparticles (DOX+CUR)@HEPT showed higher cytotoxicity than that of the free combination of (DOX+CUR), which indicated that the rationally designed carrier structure help optimize the drugs release profiles. Besides, the co-administration of both hydrophobic and hydrophilic molecules by the well-designed multifunctional micelle vehicles can be extended to other types of drugs to maximize the synergistic effect.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.