
Abstract
In this study, tricalcium silicate (C3S) calcium/polyphosphate/polyvinyl alcohol organic-inorganic self-setting composites were successfully designed. A variety of tests were conducted to characterize their self-setting properties, mechanical properties, degradation properties, and related biological properties. The composite bone cements showed a short setting time (5.5–37.5 min) with a 5:5–6:4 ratio of C3S/CPP to maintain a stable compressive strength (28 MPa). In addition, PVA effectively reduced the brittleness of the inorganic phase. Degradation experiments confirmed the sustainable surface degradation of bone cement. A maximum degradation rate of 49% was reached within 56 days, and the structure remained intact without collapse. Culturing MC3T3 cells with bone cement extracts revealed that the composite bone cements had excellent biological properties in vitro. The original extract showed a proliferation promotion effect on cells, whereas most of the other original extracts of degradable bone cements were toxic to the cells. Meanwhile, extracellular matrix mineralization and alkaline phosphatase expression showed remarkable effects on cell differentiation. In addition, a good level of adhesion of cells to the surfaces of materials was observed. Taken together, these results indicate that C3S/CPP/PVA composite bone cements have great potential in bone defect filling for fast curing.
Introduction
There is currently an increasing trend in bone defects caused by diseases and trauma. In clinical practice, it is difficult for the bone to regenerate once a wound has exceeded the “critical size defect.” 1 Therefore, the repair and regeneration of bone defects remain a serious practical challenge. For irregular defects, materials with self-setting properties are favored in surgical operations.2–5 Widespread inorganic self-setting materials include calcium phosphate bone cements (CPCs), calcium silicate bone cements, and calcium sulfate bone cements. 6 Among these, calcium phosphate bone cements have numerous advantages, including rapid solidification, injectability, favorable biocompatibility, and bone conduction. However, they have poor mechanical properties, particularly in terms of toughness and brittleness. Furthermore, hydroxyapatite, the major hydration product in CPCs, often results in unstable or unsustainable biomechanics because of its poor degradability in vivo.7,8 It is widely accepted that although bone cement has excellent degradation performance, its excessive rate of degradation leads to rapid failure at the implant site, thereby limiting its application.9,10 Calcium silicate cements not only have good bioactivity and biodegradability but also release Ca and Si ions, which stimulate the expression of osteogenesis.11,12 In addition, the relatively wide range of calcium silicate-based materials in chemical compositions may greatly influence the regulation of mechanical strength.
The basic premise of an ideal bone cement is one with mechanical properties that are adapted to a complicated mechanical environment, with new biological tissues being generated over time by degradation. 13 The degradation process is initiated by the dissolution of the material, as well as by enzymolysis, in a physiological environment.
Traditional single-component bone cements are difficult to subject to cyclic loading in clinical applications involving complicated mechanical environments. Inorganic bone cements are often combined with organic molecules, fibers, and other materials to improve their compressive strength and toughness.14–17 Excellent repair materials for bone defects should not only have outstanding biological activity to promote new bone formation but also good mechanical properties to provide support. At the same time, the proper degradation of materials is extremely necessary for quick bone repair, as it provides space for new bone growth. 18 In this study, tricalcium silicate (C3S) bone cement, which provides a stable mechanical strength, 19 was modified to obtain bone cement materials that fulfill the above-mentioned conditions for bone repair. We proposed adjusting the slow hydration and high pH of tricalcium silicate through the addition of citric acid. Citric acid can neutralize the alkalinity caused by Ca(OH)2, the hydration product, to generate calcium citrate, which can participate in bone metabolism and regulate bone density.20–23 At the same time, polyvinyl alcohol (PVA), which has rich hydroxyl groups, was also considered as a suitable additive to improve the brittleness of the inorganic phase. 24 Previous studies have found that an appropriate amount of PVA can produce strong bonds, which is beneficial to the combination of inorganic materials in bone cement and weakens the brittleness of the inorganic phase.25–27 Simultaneously, PVA increases the degradation of bone cement and hardly affects the hydration reaction, 28 calcium polyphosphate (CPP) is employed to promote the bioactivity of materials. In polyphosphate molecules, phosphate tetrahedrons are linked by oxygen bridges, with each bridging oxygen atom being shared by two tetrahedra and a calcium to phosphorus ratio of 0.5,29–31 its chain structure leads to a much higher mechanical strength than that of other calcium phosphates. 32 Polyphosphate is widely found in the osteoblasts and platelets of mammals and plays a role in storing phosphate in vivo.33,34 In an environment of excessive phosphate, stem cells can control the concentration of phosphate by synthesizing polyphosphate, thereby affecting the mineralization of the cell. 35 In a neutral pH environment, vertebrates may synthesize and release polyphosphate through alkaline phosphatase (ALP) to control the growth of apatite. 34 CPP has previously been shown to exert stronger biological activity and better cell proliferation than hydroxyapatite. 36 Moreover, polyphosphate, a synthetic metabolite polymer, has been reported to stimulate cell differentiation and mineralization.30,37–40 The biological activity and degradation performance of CPP have been significantly improved by doping with elements with smaller atomic radii, including Na, K, and Mg. 41 Therefore, CPP can enhance the biological activity and cell compatibility of bone cement and effectively control the degradation rate of bone cement.41,42
In this study, an inorganic (C3S/CPP)–organic (PVA) composite material was designed with self-solidification, based on the requirements of biodegradability, high activity, and appropriate mechanical strength of bone cement. Furthermore, a C3S/CPP/PVA composite bone cement with rapid solidification was successfully prepared, and the influence of PVA and the ratio of C3S/CPP on bone cement was explored. The setting time, mechanical strength, degradation properties, ion release, cytocompatibility, and osteogenic differentiation properties of composite bone cement were systematically evaluated.
Materials and methods
Materials
Tricalcium silicate (Ca3SiO5,C3S) was obtained from Kunshan Chinese Technology New Materials Co., Ltd. PVA (−(CH2CHOH)n−) was purchased from Shanghai Yuanye Biological Technology Co. Ltd. (Shanghai, China). Citric acid monohydrate (CA) was supplied by Chengdu Kelong Chemical Co., Ltd., Chengdu, China.
CPP was fabricated using sodium dihydrogen phosphate dehydrate (Chengdu Kelong Chemical Co. Ltd, Chengdu, China) in the laboratory. 43 Briefly, sodium dihydrogen phosphate dehydrate was heated in a corundum crucible at 800°C for 20 h to obtain sodium polyphosphate. Next, a 10% (w/v) aqueous solution of CaCl2 was added in a 2% (w/v) aqueous solution of sodium polyphosphate to form CPP gel; the CPP gel was then subsequently freeze dried to form an amorphous powder. Finally, the amorphous CPP powder was calcined at 640°C for 3 h to produce β-CPP, which was then milled and sieved (200 mesh) for application.
Preparation of cements
Based on preliminary work, the solid phases of cements were composed of C3S, CPP, and PVA powders. These powders were blended in a ball mill for 2 h at specific ratios (Table 1). The ratio of PVA was 20 wt% of the sum of C3S and β-CPP. CA was in the liquid phase, with a CA to C3S ratio of 1.5. According to preliminary work, the optimal liquid/solid ratio was 1.20 mL/g. The mixed powders and CA solution were stirred until the mixture became doughy. The resulting mixture was poured into polytetrafluoroethylene molds to obtain cylindrical specimens. After several minutes, the samples were removed and maintained in a room environment for further testing.
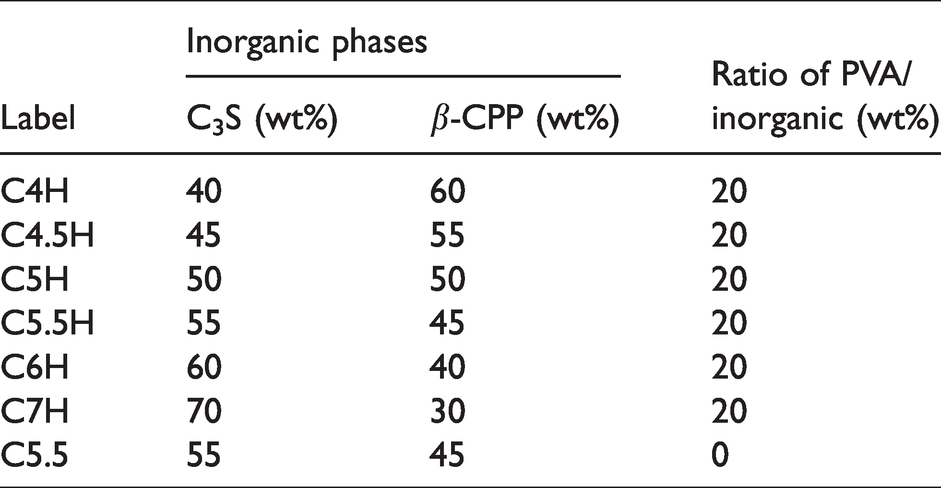
Recipe of the samples.
Microstructural characterization
Fourier transform infrared spectroscopy (FT-IR) (Nicolet PerkinElmer Co, UK) was used to characterize the functional groups of the samples. An X-ray diffractometer (XRD) (X’ Pert Pro-MPD, Netherlands) was used to identify the phases of composite cements. Scanning electron microscopy (SEM) (JEOL JSM 5600LV, Japan) was used to observe the morphology of the surface and fracture cross-section.
Setting-time and mechanical properties
The setting time of composite cements was measured using a Vicat apparatus. In line with ISO 9597–2008, the final setting time was achieved when the final setting needle of the Vicat apparatus count made an annular indentation on the surfaces of cements. The average of three samples was used as the final value.
The compressive strength was measured using a universal mechanical testing machine (RGM20; Shenzhen Reger Co., Ltd., China). The test specimens were Φ6 × 12 mm. Each value was obtained by averaging the parallel tests of five samples. Data were recorded as mean ± standard deviation (SD).
Ph value and degradation
To evaluate the degradation of cements, the cylindrical samples were immersed in phosphate buffer solution (PBS) (pH 7.40) immediately after demolding, with a solid/solution ratio of 1 g/30 mL. The immersion procedure was performed in a shaking water bath at 80 rpm at 37°C. The pH values were measured using a pH meter (PHS-3C; INESA, Shanghai, China) at immersion intervals of 1, 3, 7, 14, 21, 28, 42, and 56 days. During the first week, PBS was refreshed after every test, and every seven days thereafter. The degradation rate was assessed by the weight loss at different immersion periods (1, 3, 7, 14, 28, and 56 days). Specifically, the specimens were removed from the solution and dried at 65°C in a vacuum oven to a constant weight at the test time points. The weight loss was calculated using equation (1):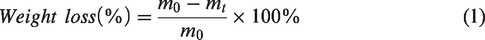
To further investigate the ion release regularity of composite cements, the released Ca, P, and Si ions were detected with inductively coupled plasma atomic emission spectrometry (ICP-AES) (iCAP7400). The degradation liquids were collected and processed with 1 mol/L HCl and diluted 20 times with deionized water for testing. Wavelengths for the elements were as follows: 317.933(II) for Ca, 213.618(II) for P, and 251.611(II) for Si. Three parallel samples were analyzed, and the results are presented as mean ± SD.
Cytocompatibility
To evaluate the biological performance of bone cements, in accordance with ISO 10993–5, the extract solutions of specimens were used for co-culture with cells, and the Cell Counting Kit-8 (CCK-8) (KeyGEN BioTECH, China) proliferation assay was employed. First, mouse preosteoblasts (MC3T3) (ATCC, Chinese Academy of Sciences, Shanghai, China) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, Thermo Fisher Scientific, Suzhou, China), supplemented with 10% (v/v) fetal calf serum (FBS) (Gibco, Thermo Fisher Scientific, Australia) and 1% (v/v) penicillin–streptomycin solution (Gibco, Life Technologies, USA), at 5% CO2 and 37°C. The cell culture medium was changed every two days. The extract solutions were prepared according to the ISO 10993–12 standard. Briefly, the specimens were immersed in DMEM at a concentration of 200 mg/mL for 24 h (37°C, 5% CO2). Then, the mixtures were centrifuged (8000 rpm, 10 min) and filtered with a 0.22 µm sterile filter. Finally, the extract solutions were diluted to different multiples (1/1, 1/2, and 1/4) with DMEM supplemented with 10% FBS. Next, 100 µL of the cell suspensions was seeded in 96-well plates containing approximately 1000 cells/well. After placing the plates at 37°C with 100% humidity and 5% CO2 for 24 h, 100 µL of different concentrations of extracts was used to replace the culture medium. The group without extract was used as the control group. After culturing for 1, 3, and 5 days, 10 µL of the CCK-8 solution was added to each well and the plates were cultured in a CO2 incubator for 4 h. Optical density (OD) was measured at 450 nm (n = 6) using a microplate reader (Multiskan FC; Thermo Fisher Scientific, Shanghai, China).
For FITC phalloidin/DAPI staining, the cells were cultured in the extracts for three days, washed with PBS, fixed with 4% formaldehyde solution for 15 min, and washed with PBS thrice. Next, 0.1% Triton X-100 (Sigma, USA) was added to the wells for permeabilization treatment for 5 min, and the wells were washed with PBS thrice. Thereafter, F-actin and nuclei were stained with FITC phalloidin (Shanghai Yisheng Biotechnology, China) for 30 min and DAPI (Shanghai Yisheng Biotechnology, China) for 5 min. Morphology of the cells was visualized using a stereomicroscope (SMZ800; Nikon) equipped with a camera (Nikon).
For cell adhesion, 1 mL of MC3T3, at a concentration of 1 × 105 cells/mL, was inoculated into the well containing the composite bone cement sample (Φ6 × 3 mm). The cells were cultured for one day at 37°C with 100% humidity and 5% CO2. After fixing with 4 vol% formaldehyde for 12 h at 4°C, the cells were dehydrated with gradient alcohol (30, 50, 60, 70, 80, 90, 95, and 100 vol%) for 10 min. Finally, SEM was used to observe cell adhesion and growth on the cement surface after freeze-drying for 2 h at ‒50°C.
Alkaline phosphatase activity (ALP)
MC3T3 (1 mL), at a concentration of 5 × 105 cells/mL, was seeded in 24-well plates at 37°C with 100% humidity and 5% CO2 for 24 h. Then, 1/1 extracts of C4H, C5.5H, C7H, and C5.5, containing osteogenic medium (1% (v/v) penicillin–streptomycin solution, 100 nM dexamethasone, 50 µg/mL ascorbic acid, and 10 mM β-glycerophosphate sodium), were replaced with growth medium, and the extracts containing osteogenic medium were changed every two days. On days 7 and 14, ALP was measured with ELISA (Shanghai MLBIO Biotechnology Co., Ltd, China) at 450 nm, and a BCA Protein Assay Kit (Solarbio, Beijing Solarbio Science & Technology Co., Ltd, China) was used to determine the total protein concentration at 562 nm (n = 6). ALP activity was calculated as the ALP level normalized to the total protein.
Alizarin red staining
Alizarin red staining was used to determine mineralization and bone differentiation in the extracellular matrix. After culturing the MC3T3 cells for 7 and 14 days, the differentiation medium was removed and the cells were washed with PBS and fixed in 4% formaldehyde for 15 min. The cells were then rinsed with PBS thrice, stained with 1% alizarin red solution for 15 min, washed with PBS thrice, and imaged under a microscope. After observation, the calcium nodules were dissolved in 0.5 mL of 10% hexadecyl pyridinium chloride solution and detected using a microplate reader at 576 nm.
Statistical analysis
Data are presented as mean ± SD. One-way analysis of variance (ANOVA) was used to compare the scores. Statistical significance was set at P < 0.05.
Results
Self-setting and mechanical properties
The setting time was affected by the use of different cements and liquid/solid ratios (Figure 1). At the same liquid/solid ratio, the setting time differed with the C3S/CPP ratio. The effect of PVA is shown in Figure 1(a). The setting time decreased as the ratio of C3S/CPP increased, and PVA prolonged the setting time when comparing C5.5 with C5.5H. Although C5.5 and C5.5H were obtained using the same inorganic recipes, the latter contained 20% PVA (Table 1). However, the setting time of C5.5 was as short as 3 min, while that of C5.5H was extended to approximately 6 min. While C4H had a longer setting time (37.5 min), C7H set after approximately 2.5 min. In addition to the C3S/CPP ratio, the liquid/solid ratio also influenced the setting time. The setting time of C4H and C6H is shown in Figure 1(b) and (c). The setting time of composite cements markedly increased with the liquid/solid ratio, which indicated that it is convenient to regulate the setting time in different situations by adjusting the liquid/solid ratio.
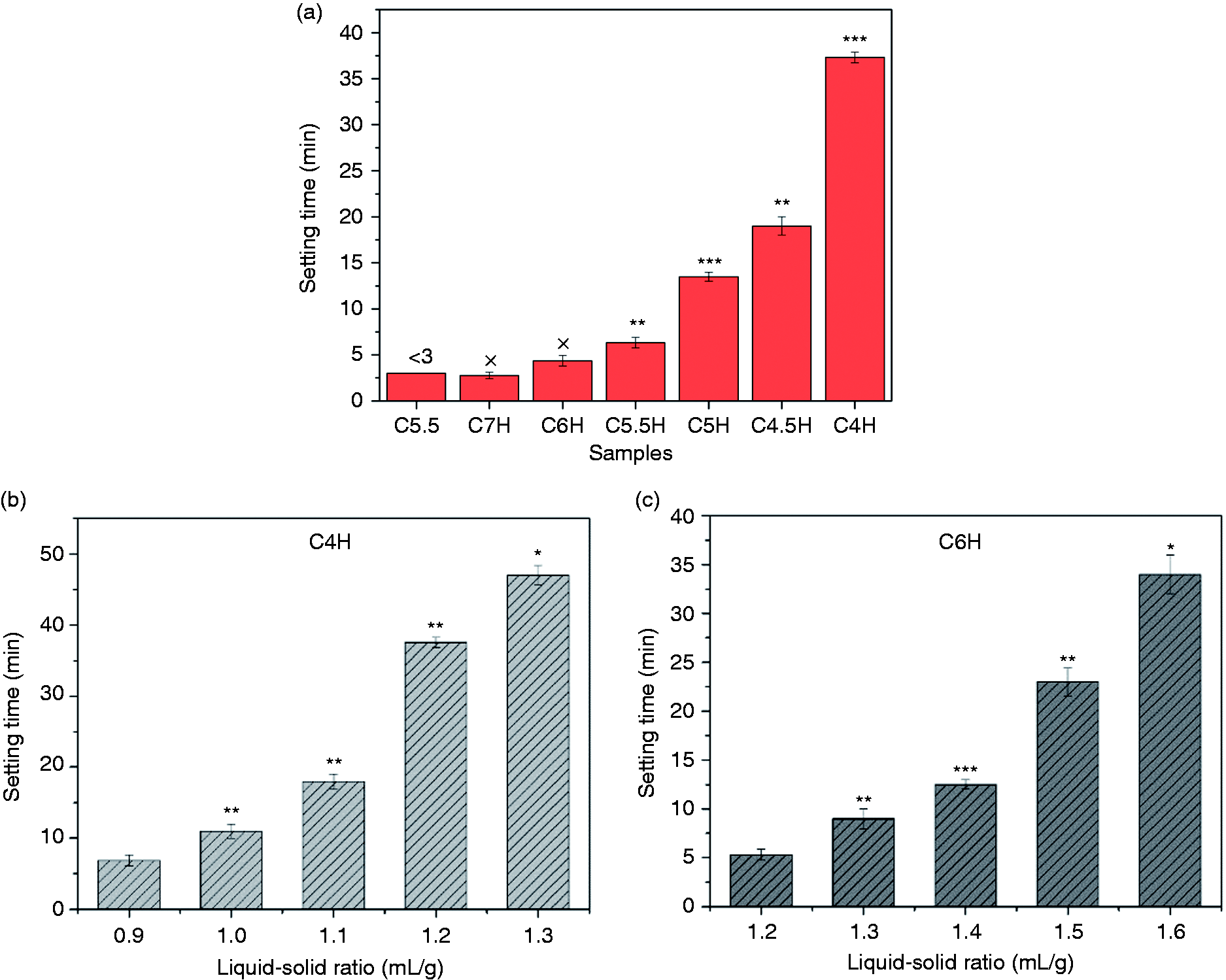
The setting time of composite cements: (a) setting time of the different samples; (b, c) setting time of C4H and C6H vs. different liquid/solid ratio. *: P < 0.05, **: P < 0.01, ***: P < 0.001; each sample was compared with the previous one (C5.5: 55% C3S and 45% CPP; C4H: 40% C3S, 60% CPP, and 20% PVA; C6H: 60% C3S, 40% CPP, and 20% PVA, and so on).
Mechanical properties of the cement are shown in Figure 2(a). A distinct increase was observed in the compressive strength from C5.5 (7.27 MPa) to C5.5H (28.46 MPa), indicating that PVA was beneficial to the improvement of mechanical properties. With an increase in the C3S content, the compressive strength of composite cements increased and was maintained at the same level (approximately 28 MPa) with a C3S/CPP of 5:5–6:4. However, the compressive strength decreased when the C3S/CPP ratio reached 7:3. Figure 2(b) shows that the addition of PVA not only remarkably improved the compressive strength but also made the cement more tenacious. C5.5H did not fail suddenly, like C5.5, after being subjected to the ultimate pressure, but slowly lost strength and was maintained at a certain level. After compression, while the C5.5H specimen maintained its shape, the C5.5 specimen cracked with the brittleness typical of an inorganic cement. The strain of the ultimate compressive strength first increased and then decreased with an increase in the ratio of C3S/CPP. The strain from C5H was the largest at 9.5%, indicating that the ratio of C3S/CPP affected the toughness of bone cement. In short, the incorporation of PVA and an appropriate C3S/CPP ratio endowed the composite cements with favorable mechanical properties.
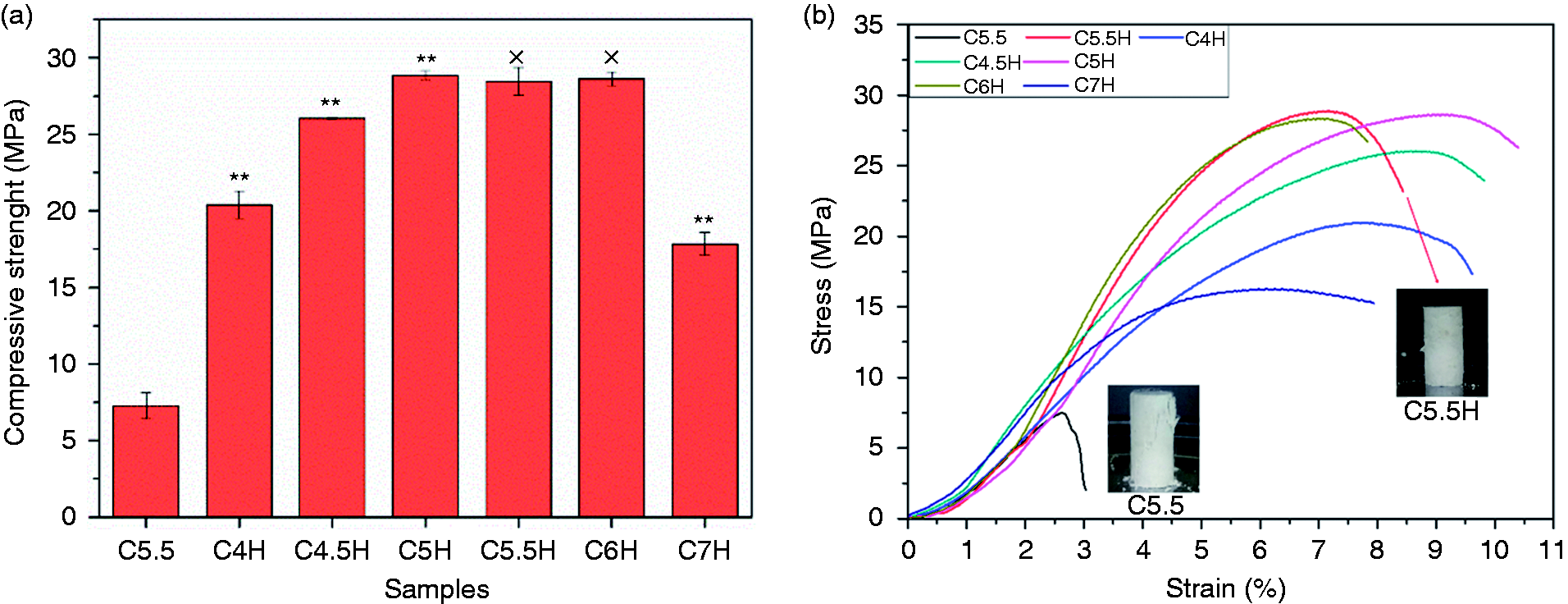
(a) Compressive strength of cements after setting for three days. (b) The stress–strain curve and digital photo (C5.5 and C5.5H) after stress. ×: P > 0.05, *: P < 0.05, **: P < 0.01; each sample was compared with the previous one (C5.5: 55% C3S and 45% CPP; C5.5H: 55% C3S, 45% CPP, and 20% PVA; C7H: 70% C3S, 30% CPP, and 20% PVA, and so on).
Component and morphology characterization
As an inorganic macromolecule, the chain length of CPP is an important characteristic. 31P-NMR of CPP was conducted, as shown in Figure 3(a). The 31P-NMR spectrum of the as-prepared CPP powder indicated that the CPP was a long-chain polyphosphate structure, and an internal phosphate peak suggested a predominant linear polyphosphate structure. The comparative average chain length (Ñ) was calculated using equation (2)44
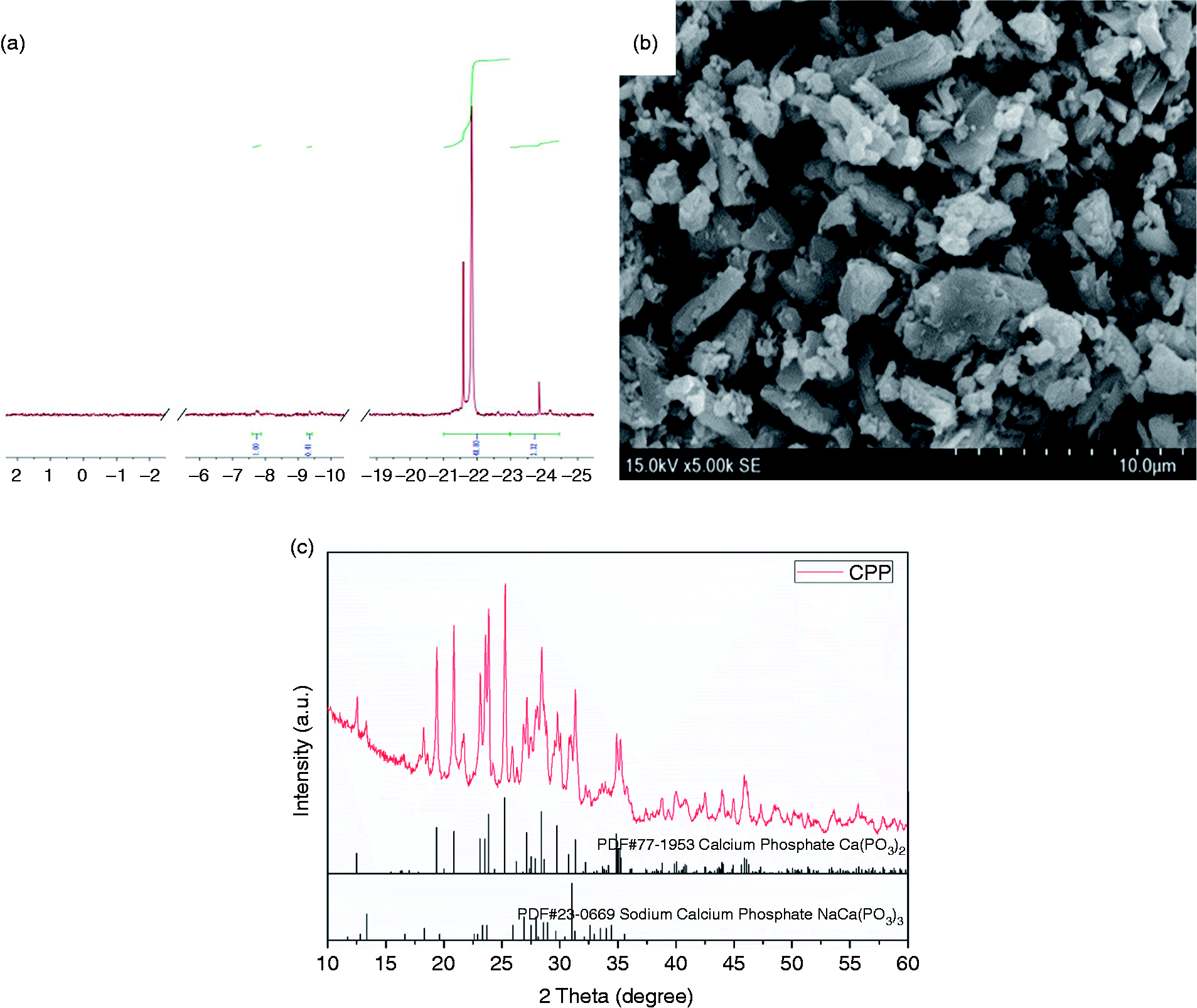
31P-NMR spectrum of CPP (a), SEM of CPP particles (b), and XRD patterns of CPP (c).
Compared to PDF 77–1953 (PDF2 2004 version), the XRD pattern of CPP (Figure 3(c)) confirmed that the main phase of the powder was β-CPP, and a small amount of calcium sodium phosphate (PDF#23–0669) existed because of the residual sodium.
The XRD patterns of the cement samples are shown in Figure 4. Similar patterns were observed for C4H, C5.5H, and C5.5, the main components of which were Ca3(C6H5O7)2·4H2O, SiO2, β-CPP, and jaffeite Ca6Si2O7(OH)6. Ca3(C6H5O7)2·4H2O was primarily attributed to the reaction of C3S and CA. The diffraction peaks of Ca3(C6H5O7)2·4H2O (PDF#01–0008) were observed at 5.9°, 22.7°, 29°, 34°, 41.2°, and 44.5°. Jaffeite Ca6Si2O7(OH)6 was formed as a result of the hydration of calcium silicate, and its peaks (PDF#46–1285) increased at 10.2°, 27.5°, and 48.7°. While the other peaks were mainly from β-CPP, peaks of calcium sodium phosphate were also observed, which came from the CPP powder. All the XRD patterns of the cements presented a number of amorphous states in the materials, which could be attributed to the PVA and the hydration products of C3S.
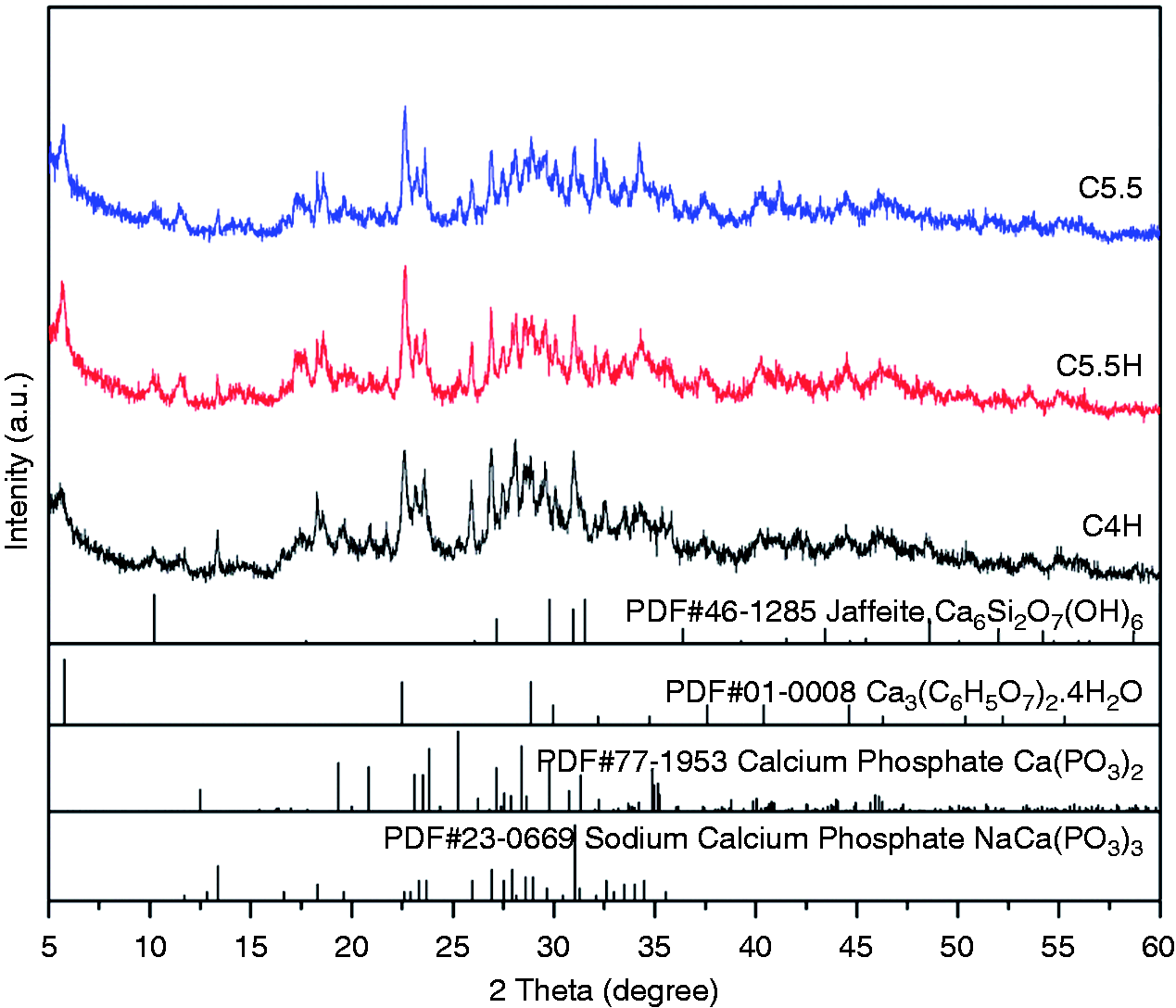
XRD patterns of composite cements (C5.5: 55% C3S and 45% CPP; C5.5H: 55% C3S, 45% CPP, and 20% PVA; C4H: 40% C3S, 60% CPP, and 20% PVA).
As shown in the FTIR spectra in Figure 5, the absorption peaks at 3421 cm−1 and 2943 cm−1 were attributed to the vibrations of –OH and C–H, respectively. As for CPP, the characteristic absorption bands at 1271, 1091, 902, and 779 cm−1 corresponded to the stretching vibration peak of P=O, the absorption vibration peak of O–P–O, the asymmetric stretching vibration peak of P–O–P, and the symmetric stretching vibration peak of P–O–P, respectively. In the spectra of C5.5 and C5.5H, with the exception of the characteristic absorption bands of CPP and PVA, newly formed absorption peaks were observed at 1671, 1540, and 1436 cm−1 (COO−) from CA, and 887 cm−1 (Si–O) and 461 cm−1 (O–Si–O) from the hydration or reaction product of C3S. In addition, intensity of the absorption bands corresponded to the content of the components in all of the composite cements.
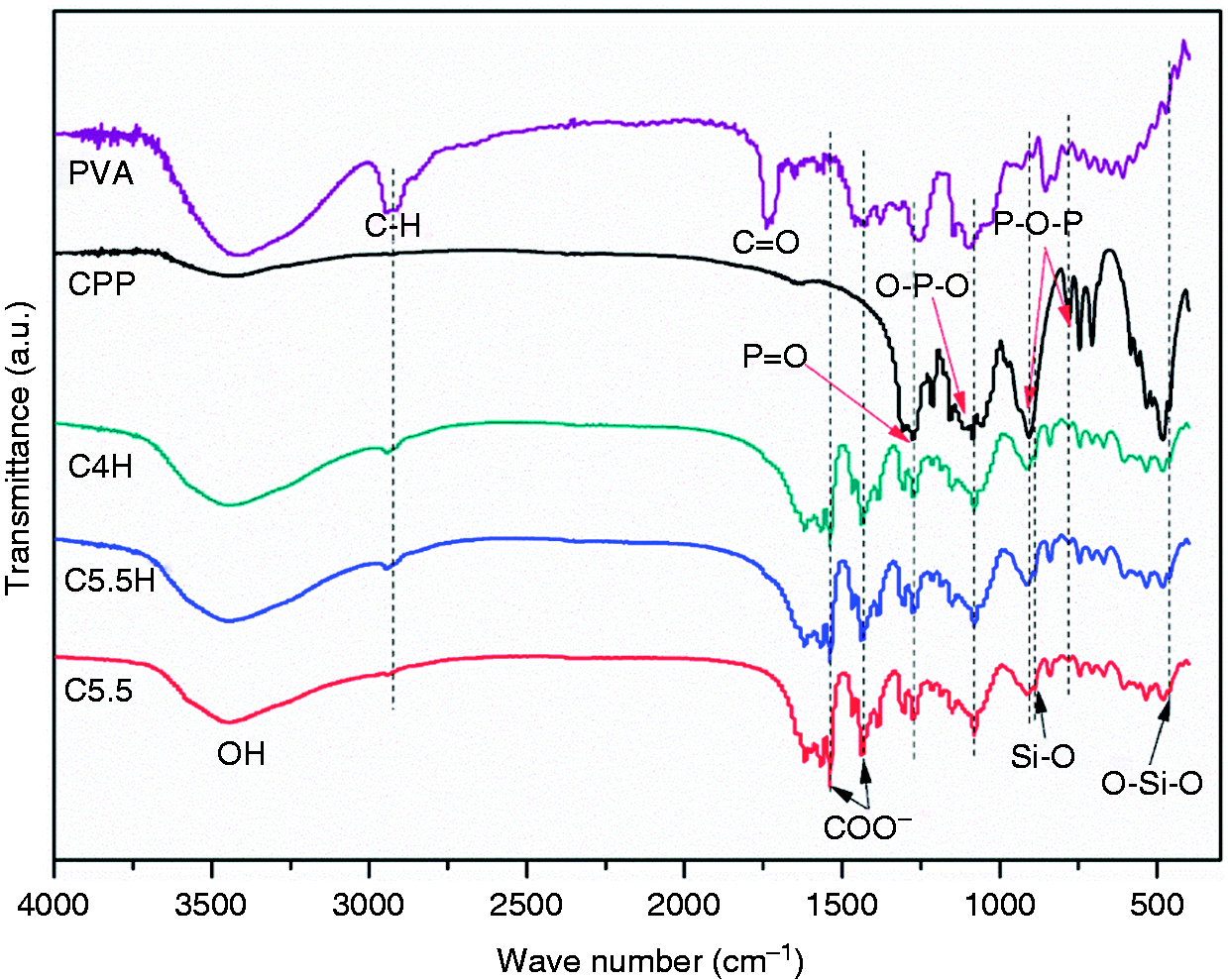
FTIR spectra of PVA, CPP, and composite cements (C5.5: 55% C3S and 45% CPP; C5.5H: 55% C3S, 45% CPP, and 20% PVA; C4H: 40% C3S, 60% CPP, and 20% PVA).
As a result of great improvement in mechanical strength after the addition of PVA, a cross-section of the materials was observed. The cross-sectional morphologies of the freeze-dried samples are shown in Figure 6. The cross-section of the cement without PVA (C5.5) showed loose plate-like overlaying. However, with the addition of PVA, the cross-sections of composite cements showed a denser structure. In addition, a number of flake crystals were interlaced with each other. Although the PVA content was the same, internal microstructures of the cements differed from those of C3S/CPP. The structure of C4H was looser than that of C5.5H, and the flake crystals in C6H were more vertical and visible than those in the others. Generally, the presence of PVA-built network of the components and the microstructures were more compact, which indicated that the organic phase dominated the arrangement of all the phases and crystal orientations, which is crucial for improving mechanical strength.

Morphologies of the cross-sections of composite cements (C5.5: 55% C3S and 45% CPP; C5.5H: 55% C3S, 45% CPP, and 20% PVA; C4H: 40% C3S, 60% CPP, and 20% PVA; C6H: 60% C3S, 40% CPP, and 20% PVA).
Degradation property in vitro
The degradation profiles in vitro after immersion in PBS at different intervals are shown in Figure 7(a). These results demonstrated that the weight loss increased continuously with the soaking time. In the first week, all samples displayed a fast degradation rate, and the weight loss of all samples was almost 15%. After the first seven days, the degradation rate of cements continued to rise slowly in the later stage of degradation. After soaking for 56 days, the weight loss of C5.5, C4H, C4.5H, C5H, C5.5H, and C6H was 27.53%, 45.12%, 49.76%, 47.15%, 46.10%, and 35.99%, respectively. Upon comparing the degradation rates of different groups, we found that the use of PVA resulted in higher rate of weight loss. Furthermore, C3S/CPP ratios were also found to affect the degradation rate at the same time. For 56 days, the degradation rates of cements supplemented with PVA were higher than those of the cements without PVA. Although C4H to C5.5H showed similar degradation rates with an increase in the content of C3S in C6H, the rate of degradation decreased. After 56 days of soaking, the samples were found to clearly retain their original shape and did not collapse (inset of Figure 7(a)), which is a characteristic of surface degradation and is conducive to clinical application. Variations in the pH of PBS after cement immersion for several periods are shown in Figure 7(b). For C5.5, without the addition of PVA, the pH value was stabilized around pH 7.1 from the beginning to the end of the experimental period. For the other composite cements, during the first seven days, the pH gradually decreased to approximately pH 6.80, at which it was maintained for five weeks, before rising to approximately pH 7 in the last two weeks. The trend in pH variation revealed that PVA influenced the degradation rate and the fluctuant of pH value. A small shift in the pH of the degradation solution was observed. However, for all composite cements, suitable pH values were observed.
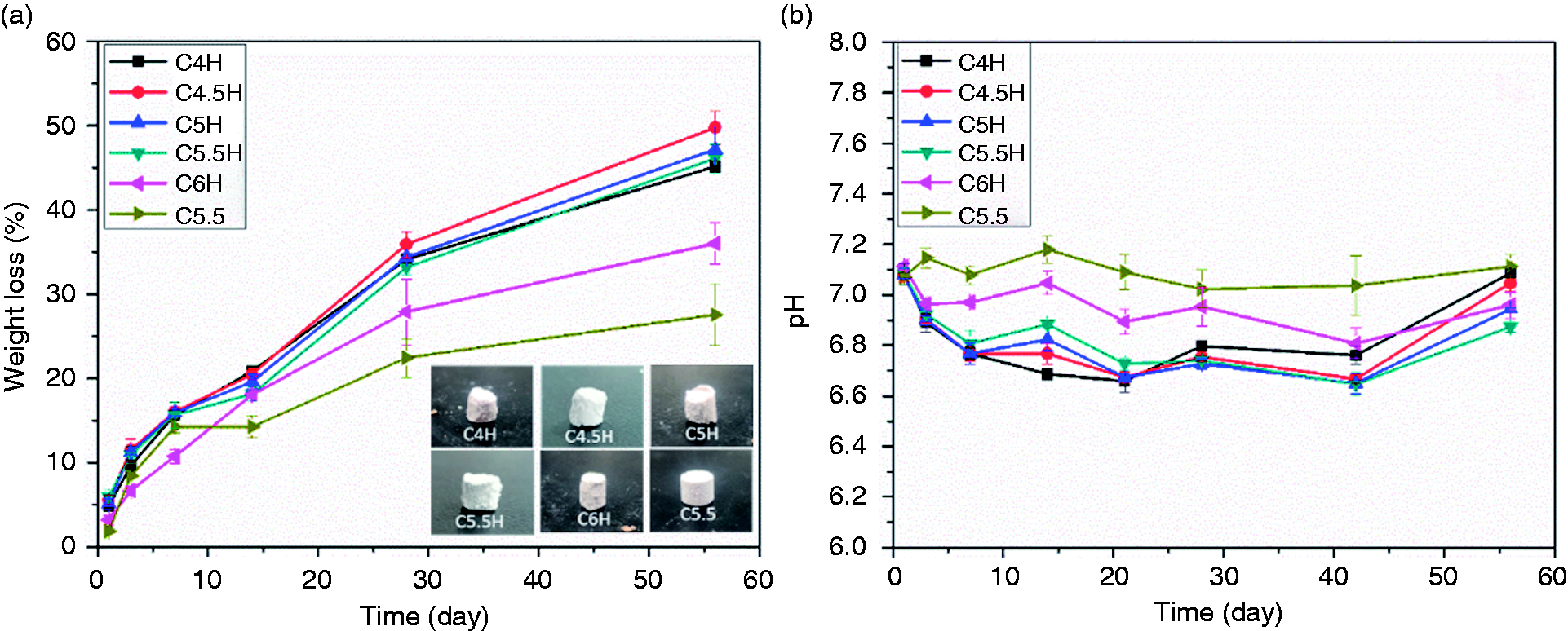
Degradation of composite cements after immersion in PBS for various time intervals; inserted images of the samples after 56 days of immersion (a) and variation in pH values over immersion time in PBS (b) (C5.5: 55% C3S and 45% CPP; C5.5H: 55% C3S, 45% CPP, and 20% PVA; C4H: 40% C3S, 60% CPP, and 20% PVA, and so on).
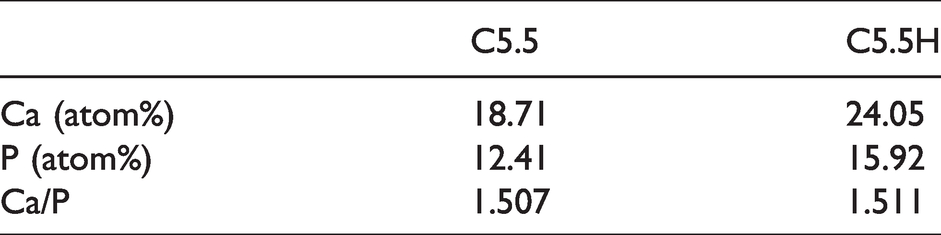
Figure 8 shows the release of Ca, Si, and P ions from C4H, C5.5H, and C5.5 at different periods after their immersion in PBS for 14 days. Ca release in the first week was labeled as Ca-7 day, Ca release in the second week as Ca-14 day, and so on. Although the composite bone cement released abundant Ca and small amounts of Si, it exhibited consumption of P in PBS. The release of Ca and Si was found to decrease with time. Compared with C5.5, the addition of PVA to C5.5 increased the ion-releasing capacity of the cement, which is consistent with the degradation phenomena. Surface morphologies of the cement samples before and after soaking in PBS for seven days are shown in Figure 9. Before immersion, flake crystals appeared piled up on the surface of C5.5, which had relatively loose surface structures compared with C5.5H and a morphology that was quite similar with the cross-sectional surface (Figure 6). After soaking, the original surface morphology disappeared and an apatite mineral layer was formed. Furthermore, the addition of PVA resulted in a change in the growth orientation of the new precipitate. The new material on the surface of C5.5 was flake-shaped, with a porous structure intertwined vertically and horizontally, while the surface of C5.5H exhibited axial growth of rod-shaped crystals arranged densely. Energy dispersive spectrometer (EDS) results indicated that elements on the surfaces of composite cement samples mainly included Ca and P after soaking in PBS for seven days. The atomic ratio of Ca/P was around 1.5 (Table 2), which is commonly associated with bone-like apatite in vitro.
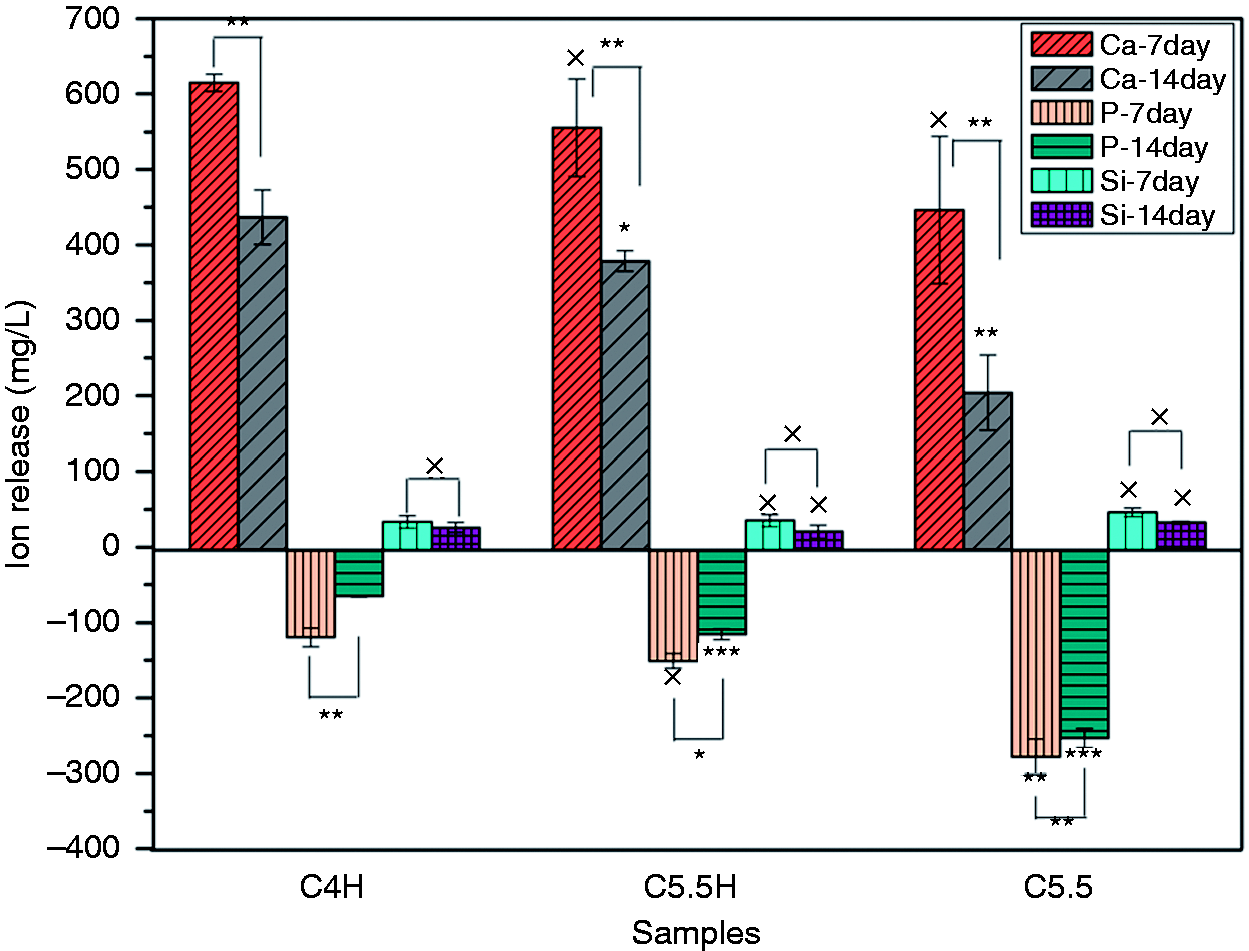
Release of Ca, Si, and P ions of C4H, C5.5H, and C5.5 at different periods of soaking in PBS. ×: P > 0.05, *: P < 0.05, **: P < 0.01; each sample was compared with the previous one and the 7-day values were compared with the 14-day values (C5.5: 55% C3S and 45% CPP; C5.5H: 55% C3S, 45% CPP, and 20% PVA; C4H: 40% C3S, 60% CPP, and 20% PVA).

Morphologies of sample surfaces before (a, c) and after (b, d) soaking in PBS for seven days: C5.5 (a, b) and C5.5H (c, d). Inserts are EDS at higher magnification (C5.5: 55% C3S and 45% CPP; C5.5H: 55% C3S, 45% CPP, and 20% PVA).
Ca/P atomic ratios after soaking in PBS for seven days.
Cell experiment
MC3T3 cells were cultured in different concentrations of cement extracts. The subsequent proliferation of MC3T3 cells was determined using the corresponding OD values, as shown in Figure 10. The OD values were found to increase gradually with culturing time. While none of the extract concentrations were detrimental to cell growth, the original extract concentration (1/1) was found to be the most beneficial to cell proliferation among different samples and culture time. The composite cement extracts with different dilution multiples showed a similar trend. In particular, on day 3, the proliferation rate with C5.5H peaked at approximately 150%. Fluorescent inverted microscope (FIM) photographs (TRITC Phalloidin/DAPI staining) of the MC3T3 cells cultured in composite cement extracts and control (complete medium) for three days (Figure 11(a)) revealed that the cells grew better in cement extracts than in the control, showing both a greater cell density and a better morphology. MC3T3 adhesion on the surfaces of composite cements, as shown in Figure 11(b), indicated that MC3T3 cells were able to spread on the surfaces of C3S/CPP/PVA and C3S/CPP composite cements, demonstrating the biocompatibility of the materials.
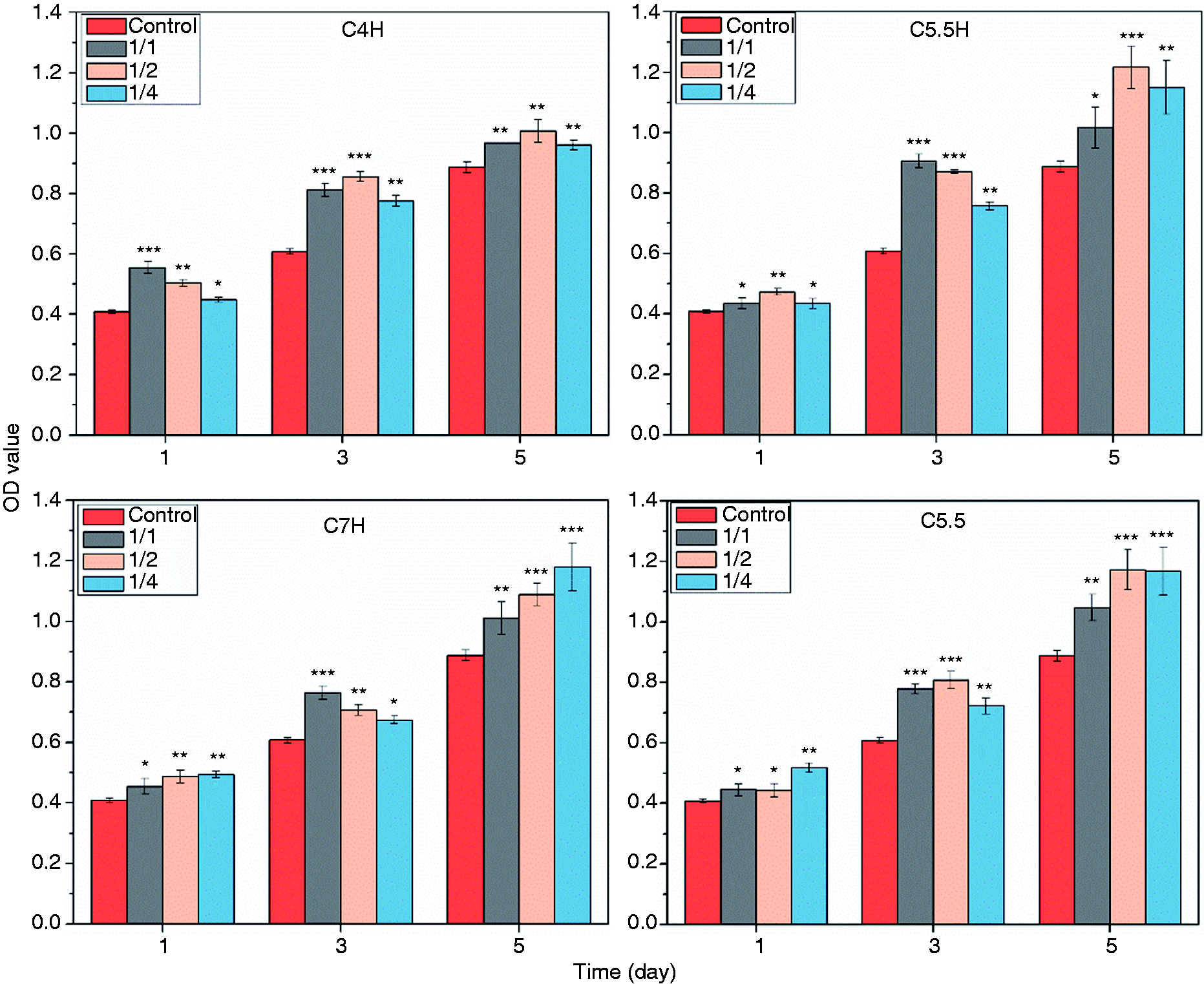
Proliferation of cells cultured with the extracts of cements with different dilution multiples (1/1, 1/2, and 1/4), as detected with CCK-8 at different intervals (1, 3, and 5 days). ****: P < 0.001, **: P < 0.01, *: P < 0.05 compared with the control group (C5.5: 55% C3S and 45% CPP; C5.5H: 55% C3S, 45% CPP, and 20% PVA; C7H:70% C3S, 30% CPP and 20% PVA).
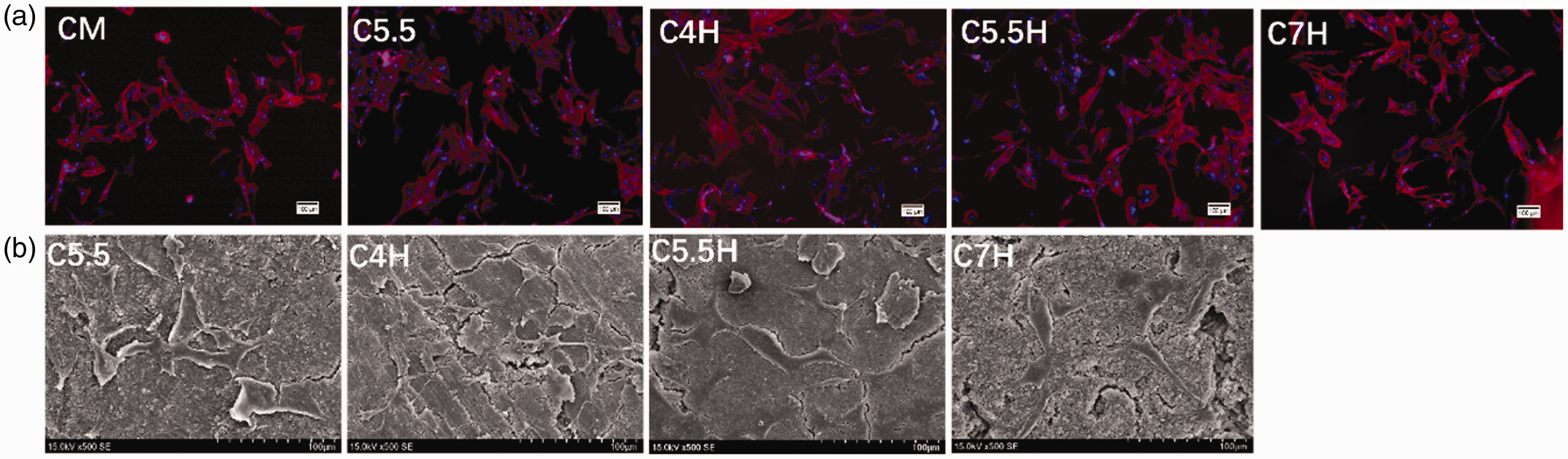
FIM photographs (TRITC Phalloidin/DAPI staining) of MC3T3 cells cultured with cement extracts and complete medium for three days (a). SEM of cell adhesion on the surfaces of C4H, C5.5H, C7H, and C5.5 composite cement samples (b) (C5.5: 55% C3S and 45% CPP; C5.5H: 55% C3S, 45% CPP, and 20% PVA; C7H:70% C3S, 30% CPP and 20% PVA).
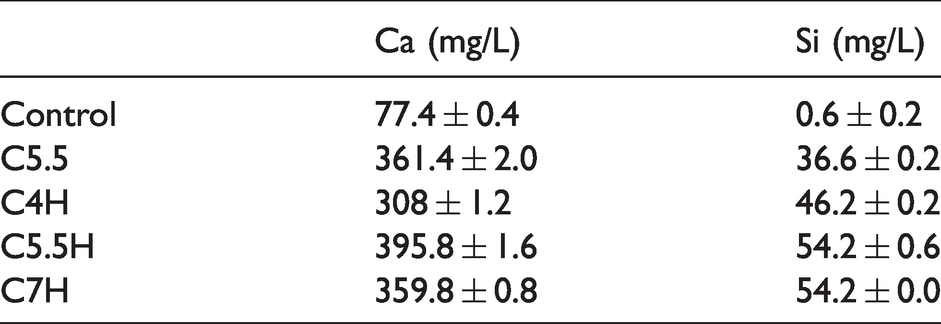
Figure 12(a) shows the Alizarin red staining results of calcification level in the extracellular matrix of MC3T3 cells after culturing at the original concentration of osteogenic extract for 7 and 14 days. On day 7, with the exception of a small number of calcium nodules in C5.5H and C7H, almost no calcium nodules were observed in the other groups. On day 14, except in the control group, a large number of calcium nodules appeared in the material groups. Combined with the semi-quantitative analysis, as shown in Figure 12(b), the calcification levels of the material groups were significantly higher than those of the control group. Among the material groups, C5.5H and C7H were found to have the highest calcification levels. As shown in Table 3, Ca and Si concentrations in sample extracts indicated that the calcification of extracellular matrix corresponded to Ca and Si ions, particularly Si ions. Figure 12(c) shows that the ALP activity of MC3T3 increased with the culture time. In particular, on day 7, the ALP activity of the material groups was significantly higher than that of the control group. A similar trend was observed on day 14; however, there was no significant difference between the C5.5 and control groups. In brief, the extracts of bone cement were conducive to the differentiation of MC3T3 cells and extracellular matrix calcification, with C5.5H being the most prominent.
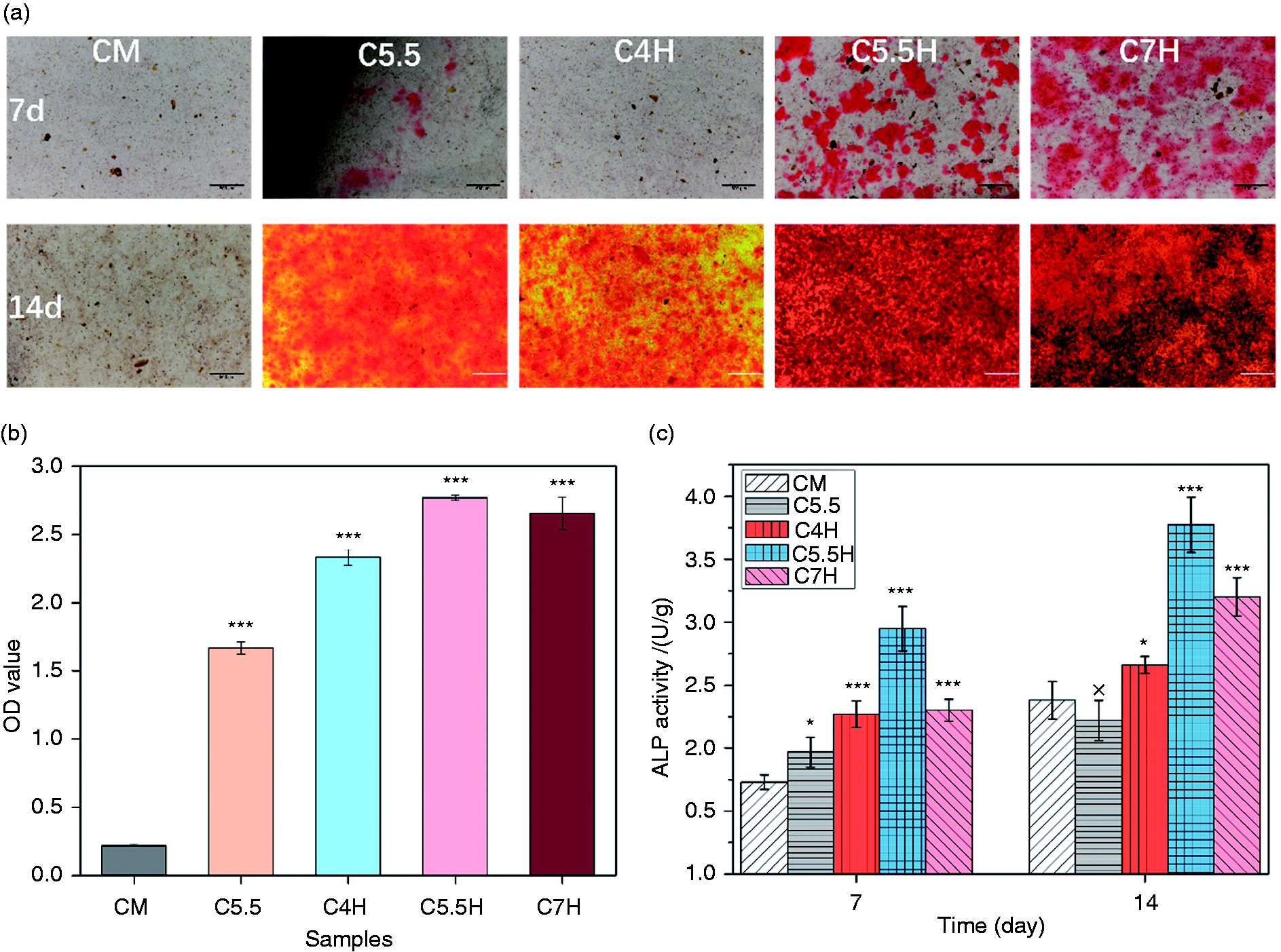
(a) Alizarin red staining of MC3T3 treated with 1/1 extract solutions of C4H, C5.5H, C7H, and C5.5 for 7 and 14 days. Osteogenic medium without the extract was set as CM. (b) Semi-quantitative analysis of the level of calcification in extracellular matrix (14 days). (c) ALP activity of MC3T3 cells treated with 1/1 extract solutions of C4H, C5.5H, C7H, and C5.5 for 7 and 14 days. *: P < 0.05, **: P < 0.01, ***: P < 0.001 compared with CM (C5.5: 55% C3S and 45% CPP; C5.5H: 55% C3S, 45% CPP, and 20% PVA; C7H:70% C3S, 30% CPP and 20% PVA).
Concentrations of Ca and Si ions in the extracts of different samples.
Discussion
Evaluating the mechanical properties, degradability, biological activity, and biocompatibility of bone repair materials is critical for their clinical application. In addition, for filling-type repair materials used in non-load-bearing defects, an excellent plasticity is one of the properties of bone fillers needed to achieve an ideal fit spatially within a complex mechanical environment for irregular defects. The focus of this study was the preparation of a degradable composite bone cement with optimal setting properties, excellent biological activity, and biocompatibility, to provide sufficient support at the defect sites.
The setting time of bone cement and the provision of sufficient mechanical support at the defect site in the early stages are important factors for evaluating bone cement. The C3S/CPP/PVA composite self-setting materials designed in this study exhibited suitable mechanical properties and pH values and adjustable setting time (Figure 1). The rapid curing of the composite bone cement was mainly due to the acid–base neutralization reaction of C3S with citric acid solution. When citric acid solution was added, C3S was hydrated and the resulting product Ca(OH)2 reacted with citric acid to form calcium citrate tetrahydrate, as confirmed from the XRD pattern. Incorporation of PVA into the composite bone cement effectively alleviated the ultra-fast solidification caused by the strong reaction of C3S and CA. Compared with the rapid solidification of C5.5, which set at 3 min, the solidification time of C5.5H extended to 6 min. This could be explained as follows: PVA, as a polyhydroxy polymer, is hydrophilic and soluble in water. The exothermic reaction of C3S and CA intensified the water absorption and dissolution of PVA, strengthened the binding of –OH of PVA to H2O, and delayed the complexation of calcium citrate with H2O. At the same time, the water-swelling PVA was effectively adsorbed on the surfaces of inorganic particles, and the –OH in the PVA formed a strong intermolecular interaction with the calcium citrate tetrahydrate by exerting an effect on –COO− and –OH, which most likely led PVA to act as a binder (Figure 13). The binder function enhances the interaction between CPP and calcium citrate. These strong interface effects led to an effective loading distribution, which significantly improved the strength and toughness of composite bone cements. As it is difficult for CPP to form a strong interface bond with calcium citrate, the mechanical strength of C5.5H was much lower than that of the PVA composite of C5.5H (Figure 2a()). These organic/inorganic composite materials exhibited compressive strength ranging from 20 to 30 MPa, which is appropriate for the replacement of cancellous bone. 46 PVA compensated for the brittleness of inorganic bone cement and increased its strength, resulting in the cement maintaining its function when subjected to complex forces during filling.
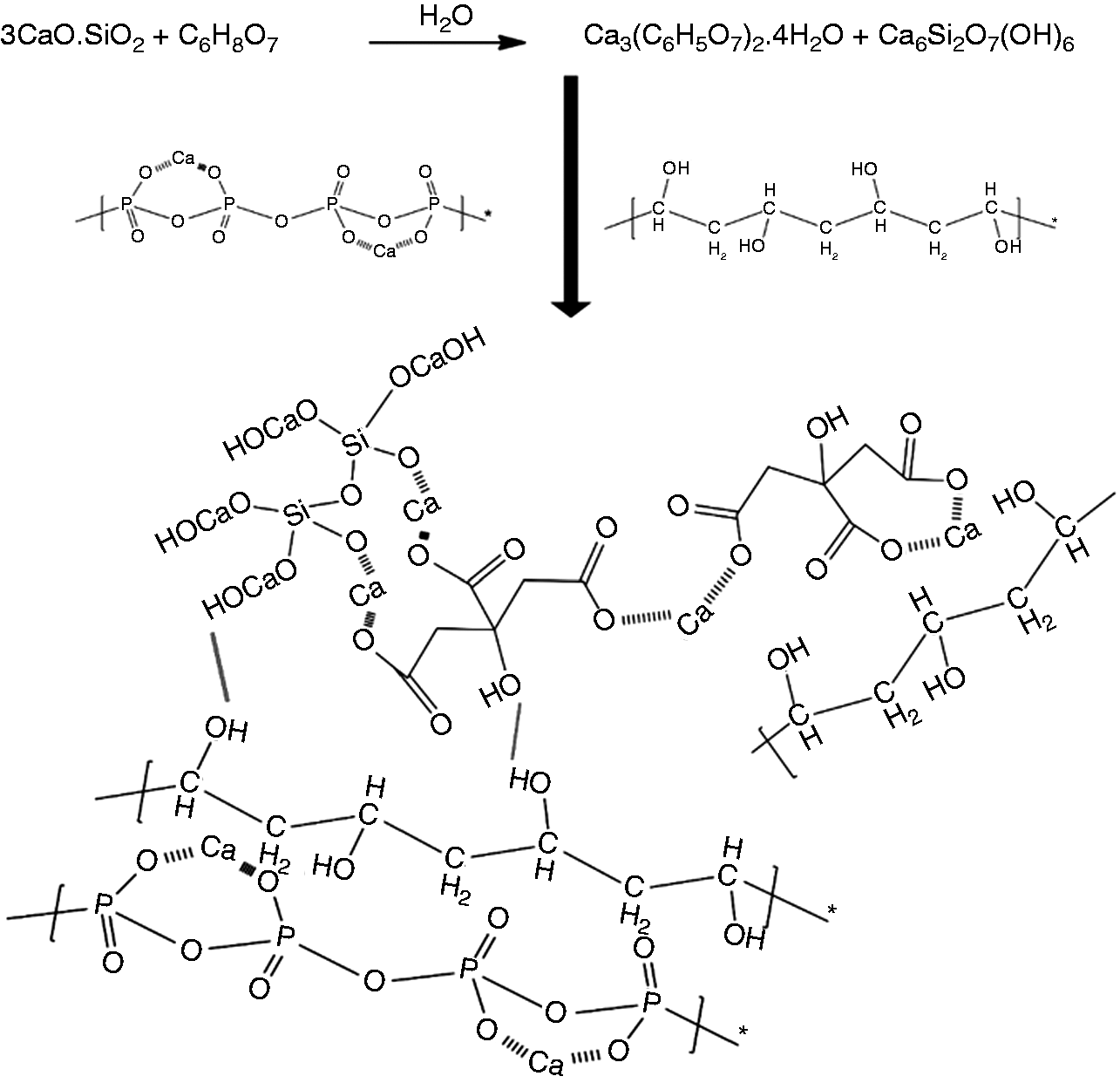
Schematic of the reinforcing mechanism of composite cements.
Degradability is an indispensable property of an ideal bone repair material. An appropriate degradation rate can provide a suitable space for new bone growth, thereby promoting the formation of an integrated bone repair tissue. Therefore, degradable materials should have sufficiently high levels of bioactivity to activate the remodeling of bones. Degradation in vivo is a very complicated process, including the processes of chemical degradation, biological degradation, and physical degradation. As a result, it is difficult to simulate the real environment in vitro. PBS only simulates ion concentration and pH environment in the body, so the in vitro degradation process of our composite bone cement should be exactly the dissolution process in PBS, including PVA dissolution, calcium citrate dissolution, calcium silicate hydrate dissolution, and CPP dissolution. As a primary study of new materials, our research focused on the properties of these materials in vitro. The C3S/CPP/PVA composite bone cement degraded to approximately 15% in the first week in vitro. A previous study reported that the composite material of HA and β-TCP shows bone formation and degradation behavior similar to that in autologous transplantation. 47 The degradation of HA and TCP mainly from TCP in the early stage, 48 approximately 14% in the first week, 49 was similar to the degradation rate of our composite bone cement. The degradation of cements was approximately 50% after eight weeks (Figure 7). During the degradation process, the materials released abundant Ca ions and some Si ions (Figure 5), which can actively stimulate bone reconstruction at the defect site. With the degradation of composite bone cement, citrate ions are continuously released, which can inhibit bone resorption and regulate bone density.22,23 Meanwhile, a Ca-P layer was deposited on the surfaces of materials during the degradation process (Figure 9). This biomineralization activity in vitro is a common prerequisite for judging the biomineralization in vivo. Formation of the Ca-P layer occurred due to two reasons: (1) due to the release of C6H5O73− by dissolution of the calcium citrate chelate with Ca2+, which resulted in the formation a positively charged complex [xC12H10Ca3O14·4H2O]y+ (1 < x < 3, 1 < y < 3), providing effective nucleation sites, 50 nucleation sites absorbing P ions to deposit apatite crystals; (2) due to the surface of the hydrated calcium silicate, where a large number of nucleation sites attract Ca2+ in the solution to interact with HPO42− and PO43− to form an apatite layer. Precisely, because the formation of the apatite layer hindered the progress of degradation to a certain extent, the release of Ca, P, and Si decreased with time. Owing to the addition of citric acid, Ca(OH)2 no longer appeared in the final product, resulting in the pH value of the suspension remaining at approximately pH 7 during the process of degradation.
To evaluate the biocompatibility of materials, observing and testing the related indicators of cells cultured with cement extracts at different time periods is an effective method. In this study, in vitro biocompatibility was evaluated based on cell proliferation, extracellular matrix calcification, and ALP activity. Although the mechanical strength of C7H was not as good as the lower content of C3S composite cement, as a systematic study on designed composite cements, the evaluation of the biocompatibility was in the range of whole recipes. The cell proliferation results indicated that none of the cements exhibited cytotoxicity, even at the original concentration of the extract. Indeed, various studies on C3S-based composite bone cement have reported different levels of cytotoxicity in the original extract and the low-fold-diluted extract, which has been attributed to the high pH value or high osmotic pressure due to the large number of dissolved ions.51–53 However, in the present study, the cells grew well in the original extract and cell adhesion experiments, which involved direct seeding of cells on the materials, further demonstrating that our materials have an excellent biocompatibility. The cell culture results (Figure 10) showed that cement extracts had the ability to promote cell proliferation at different time points; however, varying effects were associated with different dilutions. It is well known that Ca and Si ions in the extract stimulate cell proliferation and play a vital role in cell differentiation and extracellular matrix mineralization.54,55 At the same time, ion concentration in the extract significantly affected cell growth and reproduction. In this study, although all the concentrated extracts showed beneficial effects on cell proliferation, the low-fold dilution of the extract was found to have a significantly higher promotion effect on cell proliferation than the high-fold dilution because of the lower concentration of Ca and Si ions. Compared with that in the control group, the Ca and Si ions of the composite bone cement (Table 3) were found to be beneficial for stimulating the proliferation of MC3T3 cells and the mineralization of extracellular matrix. The content of Ca and Si ions in C5.5H was higher than that in other samples, and it showed a better proliferation effect and a higher ALP activity, consistent with the results of a previous study. Adhesion of MC3T3 cells on the surface of C3S/CPP/PVA composite is shown in Figure 12. The cells were spread into a flat shape and obvious filopodia could be observed in many places. Figure 11 shows the morphology of MC3T3 cells cultured for three days. Compared with that in the control group, the cells spread more in the composite cement extracts and grew more antennae, which was consistent with the cell proliferation results. Evaluation of extracellular matrix mineralization and ALP activity was mainly based on the formation of calcium nodules and the related protein expression of MC3T3 cultured in osteogenic medium for different time intervals. Previous studies reported that the expression of MC3T3 cell differentiation was not marked in the early stage, 56 with extracellular matrix mineralization and alkaline phosphatase expression observed after nine days. 57 Compared with those in previous studies,58–60 the composite cements prepared in this study were effective in stimulating the differentiation of MC3T3 cells. Figure 12 shows that a large number of calcium nodules were formed on day 14, and their number was significantly (P < 0.05) higher in the material groups than in the control group. The PVA-containing composite materials were higher than the inorganic cement (C5.5). The results showed that Ca and Si ions derived from composite bone cement contributed to the mineralization of extracellular matrix. ALP activity is an important indicator of the emergence of osteoblasts, which indicates the successful differentiation of MC3T3 cells. ALP activity increased with culture time, and the number of material groups was higher than that of the control group on days 7 and 14 (Figure 12). Taken together, the proliferation, adhesion, extracellular matrix mineralization, and ALP activity of MC3T3 cells indicated that C3S/CPP/PVA composite bone cements had excellent biological properties in vitro, in particular the component C5.5H, which showed suitable properties for fixing and accelerating the repair of defects.
Conclusions
In this study, degradable C3S/CPP/PVA composite cements were successfully fabricated for the first time. The cements had an adjustable setting time, which could be tailored in the range of 5.5–37.5 min. The composite cements with 20% PVA in the inorganics displayed an appropriate compressive strength of approximately 28 MPa when the ratio of C3S/CPP in the inorganics was 5:5–6:4, as well as suitable degradation rates in the range of 36–49.7% in PBS after eight weeks of immersion. With regards to the biological properties of composite cement in vitro, cell experiments demonstrated that all concentrations of the C3S/CPP/PVA composite cement extract promoted cell proliferation and were beneficial to the differentiation and extracellular matrix calcification of MC3T3 cells. In addition, MC3T3 cells adhered well to the surface of composite bone cement. Combined with these preliminary results, the C3S/CPP/PVA cement series appears to be a promising bone-filling material. Among these, the C5.5H recipe exhibited the most remarkable bioactivity.
Highlights
Organic-inorganic composite bone cement was designed for fast curing and a suitable pH. PVA significantly improved the mechanic strength and deformation displacement of C3S/CPP cement. Composite cements showed excellent biocompatibility and stimulated the proliferation and differentiation of MC3T3.
Footnotes
Acknowledgements
We would like to thank the Analytical & Testing Centre of Sichuan University for FTIR, SEM, and XRD characterization. We would also like to thank Ms. Xu Hou for her help with measurements using the flame atomic spectrophotometer.
Declaration of conflicting interests
The author(s) declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclose receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (51773123).