
Abstract
Graft substitute is a mature treatment tool in craniofacial bone repair. However, stress shielding and immutability of structure limit its use in patients with congenital defects. Therefore, a regenerative graft would be best suited for repair. Mesenchymal stem cells (MSCs) have been shown to be feasible in regenerative medicine and the clinical treatment of bone repair. The aim of this study was to propose a strategy that would directly blend graphene oxide (GO) and MSCs with gelatin methacrylate anhydride (GelMA), as bioink, to generate the scaffold for bone regenerative repair. The survival and osteogenic capacity of MSCs in the composite bioink were assessed by cell viability and proliferation assays, along with expression analysis of osteogenesis-related genes and proteins, and targeted immunofluorescence. The introduction of GO to the printing process had no influence on cell printing, viability, or printability of GelMa. However, the GO-involved structure exhibited a positive influence on MSC proliferation, without significantly affecting cell viability. Alkaline phosphatase was expressed more in cells cultured with GO than in those with pure GelMA. In addition, GO promoted the expression of osteogenesis-related genes and proteins, such as osteopontin, osteocalcin, and RUNX2. Collectively, the composite bioink enhanced cell proliferation and adhesion, as well as osteogenic differentiation properties, compared with pure GelMA.
Keywords
Introduction
The large number of craniomaxillofacial bone repairs conducted in recent years has revealed unique features of the engineered tissue, including better mechanical properties, higher porosity, biocompatibility, and biosafety, compared to those of other tissues.1,2 Therefore, biological scaffolds for craniomaxillofacial reconstruction should have sufficient structural stability to counteract the contraction force and provide a suitable environment for subsequent tissue growth. Bioinks are special suspensions, consisting mainly of hydrogels that encase cells or tissues, mimicking the natural extracellular matrix, to provide a microenvironment for cell growth and ensure water retention within cells. Thus, bioinks need to be produced in ways that guarantee specific properties, such as biocompatibility, biodegradability, and appropriate mechanical properties.
The classical materials currently available include synthetic, natural, and composite materials. Synthetic polymers such as polyglycolic acid, polylactic acid, and polycaprolactone have good printing resolution and mechanical strength. Zheng et al. 3 mixed silk protein with polyethylene glycol to make a bioink in which human bone marrow mesenchymal stem cells could survive for a long time. Although synthetic polymer materials have advantages, they lack osteoinductivity and cannot induce osteoblast differentiation or promote bone regeneration; therefore, 3D bioprinting is often performed using synthetic materials combined with inorganic materials. Natural polymer materials include collagen and gelatin. Natural hydrogels are preferred for wrapping cells because of their hydrophilic properties and similarity to extracellular matrix components.4,5 Zhai et al. 6 wrapped primary rat osteoblasts with sodium hyaluronate for bioprinting and UV crosslinking for 30 min did not reveal cell damage. Another research team printed cells wrapped in a natural polymer material simultaneously with the scaffold material and the cells survived on the scaffold at higher temperatures.7,8 Wrapping the cells with natural polymer materials helps to protect them during printing and cross-linking. In several studies, various natural polymer materials were mixed with cells to prepare bioinks making use of the chemical properties of different materials. Gelatin combined with sodium alginate is the most used association; it allows the preparation of hydrogels with certain mechanical properties by applying the chemical cross-linking properties of alginate and the thermal sensitivity of gelatin. 9
Gelatin methacrylate anhydride (GelMA) is synthesized via a direct reaction of gelatin and methacrylic anhydride in phosphate buffer at 50°C. It is a photosensitive biohydrogel material that can form a three-dimensional (3D) structure suitable for cell growth and differentiation, and has a certain strength owing to a curing reaction. 10 Preservation of structural integrity and mechanical strength are the two most important criteria in bioprinting, and GelMA not only meets these requirements but also has good biocompatibility, low immunoreactivity, and potential presence of biologically active motifs in its chemical structure. However, low concentrations of GelMA bioinks result in low mechanical strength and fidelity. Although higher concentrations of bioinks can overcome such limitations, they inhibit cell viability and the development of normal tissue morphology. Therefore, designing low-density GelMA bioinks with better mechanical properties, scaffold fidelity, and cellular function remains a challenge. The addition of polymers and nanomaterials to aqueous GelMA bioink solutions improves the printing performance, cell viability, proliferation, and differentiation, as well as the mechanical and chemical stability of the printed tissue under organismal conditions. Some results have shown that graphene oxide (GO) enhanced the mechanical properties and improved the printability and shape integrity of 3D printed structures compared with GelMA alone.11-13 The enhanced mechanical properties of GO are supposed to be due to the covalent interaction between GO and methacrylate groups on the GelMA chain. 12
Graphene oxide exhibits various functionalities, and has promising applications in the biomedical field owing to its unique structure and a surface rich in functional groups. The functional groups help disperse the GO particles in solvents and act as crosslinks between polymers and polymer-biomolecule bonds. 14 Compared to carbon nanotubes, GO can disperse more easily in solvents and form stable colloidal suspensions owing to the formation of hydrogen bonds between its surface functional groups and water molecules. Previous studies had shown that titanium discs coated with a GO monolayer can promote cell proliferation, 15 and that GO can improve the mechanical strength of ultra-high-molecular-weight polyethylene. 16 Furthermore, GO can improve cell adhesion, proliferation, and growth, as well as osteogenic differentiation of mesenchymal stem cells (MSCs).17-20 Several studies have explored the features of GO when incorporated into different hydrogel scaffolds, such as alginate,21,22 collagen, 23 and GelMA,13,24 and when forming 3D structures by crosslinking with polymer chain scaffolds. The aim of the current study was to examine whether the GelMA/GO hydrogel could successfully promote osteogenesis.
Materials and methods
Isolation, culture, and differentiation potential of MSCs
Adipose tissue was obtained from the abdomen or thighs of individuals aged 20–29 years, who had undergone liposuction for localized obesity. The procedures were approved by [details omitted for double-anonymized peer review]. Freshly aspirated adipose tissue was added to an equal volume of phosphate-buffered saline (PBS), mixed well, and centrifuged at 500 ×g for 3 min. The lower hematocrit layer was removed therefrom. This was repeated several times until the hematocrit layer had a light pink color. The fat pellet was added to 0.4% type I collagenase and shaken for 1 h at 200 ×g and 37°C in a horizontal shaker. Thereafter, the samples were centrifuged at 200 ×g for 5 min, the upper lipid margin removed, Dulbecco’s PBS added, the samples mixed by turning the tubes upside down, and finally the samples centrifuged at 200 ×g for 5 min. α-Minimum Essential Medium (MEM, 10 mL) was added next, and the cells were seeded on 10-cm cell culture dishes (Thermo Fisher Scientific, Waltham, MA, USA) by blowing them apart. The dishes were incubated in a 5% CO2 cell culture chamber for 72 h at 37°C. Adherent cells were considered to be primary adipose MSCs. The MSCs were cultured in α-MEM (Gibco, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin.
Subsequently, 2 × 105 cells were cultured in 6-well plates, and the osteogenic, lipogenic, or chondrogenic medium was changed every 2 days when cell confluence reached 70%. The osteogenic differentiation medium was prepared in α-MEM supplemented with 10% FBS (Gibco), 50 μg/mL ascorbic acid (Solarbio, Beijing, China), 100 nM dexamethasone (Solarbio), 10 mM β-glyceraldehyde-3-phosphate (Sigma Aldrich, St. Louis, MO, USA), and 1% antibiotic/antimycotic (Gibco). After 21 days of differentiation, lipogenic cells were stained with Oil Red O (Solarbio) working solution, chondrosphere sections were stained with Alcian Blue (Oricell, Suzhou, China) working solution, and osteogenic cells were stained with Alizarin Red (Oricell). The induction effect was observed and imaged using an inverted microscope (Carl Zeiss, Oberkochen, Germany).
Flow cytometry analysis
Flow cytometry was conducted to detect MSC surface markers. Cells were suspended in PBS at a density of 5 × 106 cells/mL. Nine aliquots containing 100 μL of the cell suspension were prepared. Antibodies were added using the human BD Stemflow MSC Analysis kit (BD Biosciences, Franklin Lakes, NJ, USA) and incubated at 4°C in the dark for 30 min. Flow analysis solution (1 mL) was added to each tube, which was centrifuged at 400 ×g for 3 min thereafter. Next, the cells were resuspended in 500 μL of flow analysis solution and analyzed on a BD FACSAria II cell sorter (BD Biosciences). Cells from the 3rd–5th generation were used for the experiment.
Preparation of GelMA/GO Bioinks
Compositions of the bioinks.

Cell printing
Cells were mixed with GelMA or GelMA/GO solution at a cell density of 2 × 106 cells/mL. A sterile syringe was filled with the bioink and cooled to 20°C. Bioinks require approximately 130–150 kPa of air pressure to squeeze through the 260-μm nozzle. Printing was performed at a rate of 8 mm/s and the scaffolds were cross-linked using UV irradiation for 30 s. Dimensions of the solid cube constructs were 10 × 10 × 2.5 mm3. The cells were cultured in α-MEM (Gibco), which was replaced by osteogenic differentiation medium 3 days later. During further osteogenic culture, the new osteogenic medium was changed every 2 days. The overall structure of the hydrogel scaffolds was observed using an inverted microscope.
Cell viability and area
To evaluate cell viability, the scaffolds cultured in vitro for 1, 7, 14, and 21 days were subjected to live/dead staining tests. Calcein AM and propidium iodide (Invitrogen, Waltham, MA, USA) were mixed in a 2:1 ratio in PBS, added to the constructs, and incubated in the dark for 10 min. Photographs were obtained for live and dead cell identification, and counting was performed using a laser confocal microscope with the FIJI software (https://imagej.net/software/fiji/). Three sample sets were used for each time point in each group.
Cell proliferation
The Cell Counting Kit (CCK)-8 assay was used to examine the proliferation of MSCs in the scaffolds. Three to five scaffolds were collected on 1, 7, 14, and 21 days. A working solution (α-MEM:CCK-8, 10:1) was added to each well under light-proof conditions. The working solution was incubated at 37°C for 2 h, transferred to a 96-well plate at 100 μL per well, and the respective absorbance at 450 nm was determined using a full-wavelength detector (Thermo Fisher Scientific, Waltham, MA, USA).
Alkaline phosphatase (ALP) activity assay
Osteogenic differentiation of MSCs in bioprinted scaffolds was assayed on days 7, 14, and 21 according to the ALP assay kit instructions (Beyotime, Shanghai, China). The scaffolds were placed in a homogenizer with 100 μL of 1% TritonX-100-lysed cells and, following lysis, the liquid was collected in Eppendorf tubes and centrifuged at 12,000 ×g for 5 min at 4°C. Normalized ALP activity (IU/mL) was calculated according to the BCA protein assay kit (Thermo Fisher Scientific) and ALP kit instructions. Three samples were analyzed from each group, and three ALP and BCA assays were performed for each substrate.
Cell cytoskeletal organization analysis
The cytoskeleton was evaluated by immunofluorescence staining. The specimens were fixed in 4% formaldehyde for 30 min and soaked in Triton X-100 (0.1%) for 15 min. A drop of PBS solution containing 1% (w/v) bovine serum albumin was added to the sample and incubated for 30 min. Next, the specimens were incubated with Alexa Fluor-488-tagged phalloidin for 1 h for F-actin detection. Finally, the samples were stained with 4′,6-diamidino-2-phenylindole (Yeasen, Shanghai, China). Imaging was performed using an Eclipse Ti confocal microscope (Nikon, Tokyo, Japan) to obtain 3D render images.
Quantitative real time-polymerase chain reaction (qRT-PCR) analysis
Primer sequences used for RT-PCR of bioprinted constructs.

Western blot
Samples for protein analyses were collected on days 14 and 21. Total protein was extracted by adding 200 μL of RIPA buffer (Beyotime) and the lysate was sonicated. Total protein samples (30 μg) were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes. The membrane was incubated for 8 h at 4°C with antibodies targeting GAPDH (1:1,000; ab8245), osteocalcin (OCN) (1:1,000; ab133612), RUNX family transcription factor 2 (Runx2) (1:1,000; ab192256), and osteopontin (OPN) (1:1,000; ab214050), all purchased from Abcam (Cambridge, UK). Next, it was incubated with horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse secondary antibodies (1:1,000; SA00001-2 and SA00001-1, respectively; ProteinTech, Rosemont, IL, USA) for 1 h at 25°C. The film was laid flat, ECL luminescent solution (#29050; Engreen, Beijing, China) was added, and eventually, the bands were detected in a dark room.
Statistical analysis
All experiments were repeated thrice. Statistical analysis was performed using SPSS version 20.0 (IBM Corp., Armonk, NY, USA). Two-way analysis of variance was performed, followed by Tukey’s post-hoc test. All data are expressed as mean ± standard deviation of the mean (SD). Differences were considered significant at p < 0.05.
Results
Figure 1 shows the P2 generation of MSCs. The latter were cultured in osteogenic, chondrogenic, and lipogenic induction solutions for 21 days and were stained with (Figure 1(b)) Alizarin Red, (Figure 1(c)) Alcian Blue, and (Figure 1(d)) Oil Red O, respectively. Flow cytometric analysis of phenotypic markers in MSCs showed that 98.3%, 100.0%, and 99.8% of the cells were positive for CD105, CD73, and CD90, respectively. MSCs were negative for CD34, CD11b, CD19, and CD45 (Figure 1(e)). The results confirmed that the cells that were harvested were MSCs. Figure 2 shows the overall structure of the hydrogel scaffolds, observed under an inverted microscope. In-vitro culture and differentiation of MSCs. ( Overall structure of the hydrogel scaffolds seen under an inverted microscope. Images of GelMA alone and GelMA/GO scaffolds ((a), (b), (c), and (d) represent 0GO, 0.5GO, 1G, and 2GO, respectively).

Next, MSCs survival on days 0, 3, 7, 14, and 21, after bioprinting, was evaluated (Figure 3(a) and (b)). For all groups, most MSCs were alive, and only a few dead cells were detected at each time point. Live/dead staining was performed 3 h after printing, and the cell viability exceeded 95%, being not significantly different across groups, indicating the printing process to have less cell-damaging effects. The fluorescence intensity of the four different GO groups was lower than that of the 0GO group, which may be related to the fluorescence blocking effect of GO powder. On day 7, cell stretching and increased projection area were observed, and the blocking effect of GO on fluorescence was diminished. Furthermore, the results from day 21 showed the GO group to have larger and more widely distributed cells, with increased cell projection area than in the 0GO group. The result indicated that GO increases cell-cell interaction. Next, the proliferation rate of cells was evaluated by the CCK-8 assay (Figure 3(c)). The pure GelMA+hADSC-loaded scaffold showed the earliest proliferation peak compared to the other groups, whereas cells in the hydrogel with GO exhibited a longer proliferation period (1GO group showed the fastest proliferation rate at the beginning of day 9 of culture). Similar proliferation rates were observed in all groups on day 21. Impact of the bioprinting process on viability and proliferation of MSCs. (a) Live/dead staining of MSCs in bioprinted scaffolds on days 1, 7, 14, and 21. (b) Quantification of cell viability, shown as percentage (%). (c) Proliferation of MSCs determined by CCK-8 assay; n = 3 per group, all data expressed as mean ± SD.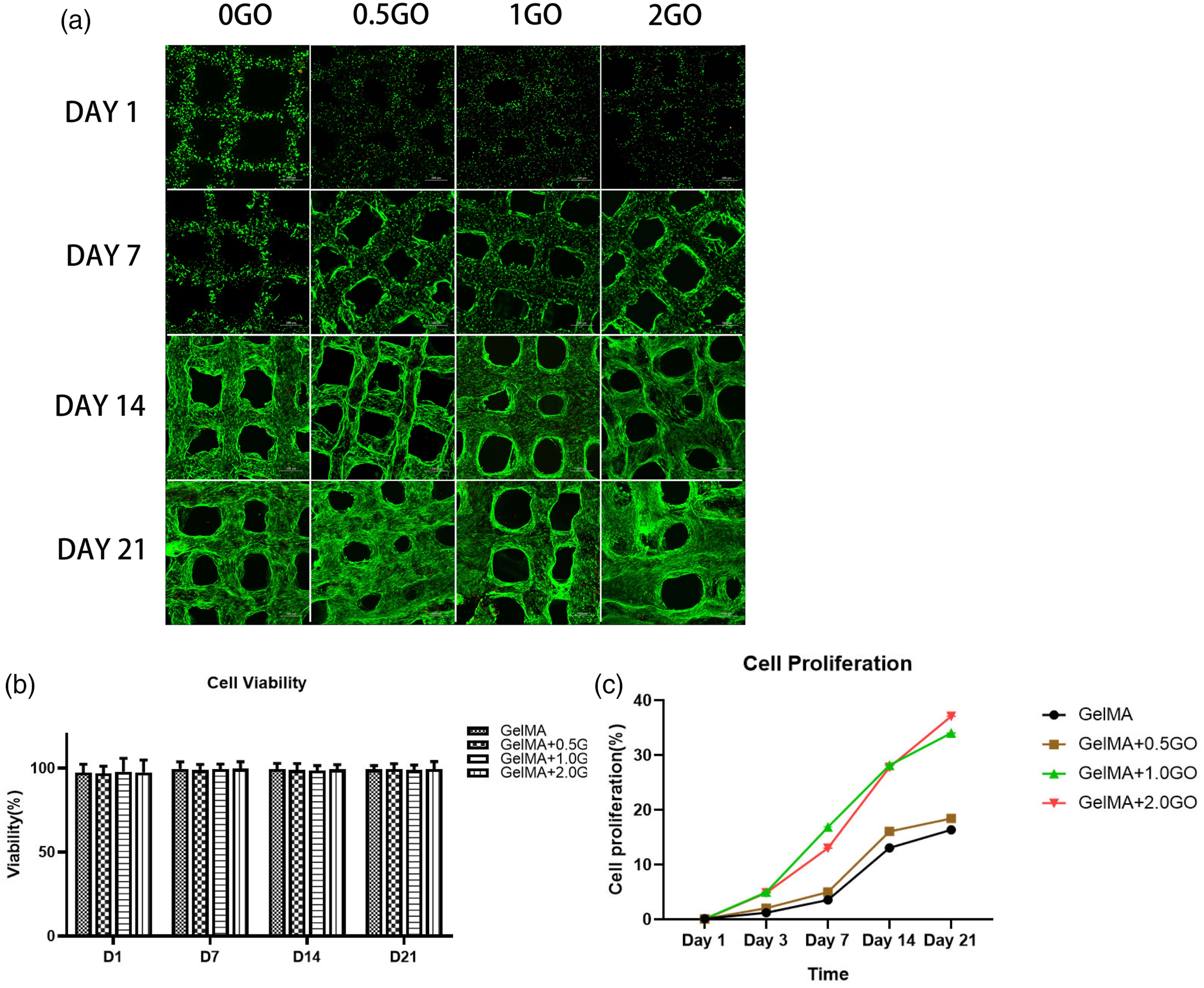
Immunofluorescence analysis of F-actin fibers and fibronectin in MSCs contained in the scaffolds further showed that, although 1GO and 2GO groups had more green (F-actin) fluorescence, and the signal intensity increased within days 14–21 (Figure 4(a) and (b)), semi-quantitative expression of F-actin was the highest in the 2GO group (Figure 4(b)). Moreover, assessment of ALP activity (Figure 4(c)), which indicates the presence of osteoblast cells, showed that it increased with time in each group, being significantly higher in the 1GO and 2GO groups after 14 days of culture. After 21 days of culture, ALP activity was increased in the 0GO group than that after 14 days, indicating that more cells were maintained in the osteogenic progenitor stage. In addition, ALP activity was lower in the 1GO group than in the 2GO group. Taken together, the results suggested that MSCs secrete more F-actin fibers during the differentiation process in response to GO, thereby eventually promoting osteogenesis. Assessment of the overall presence of MSCs in the scaffolds. (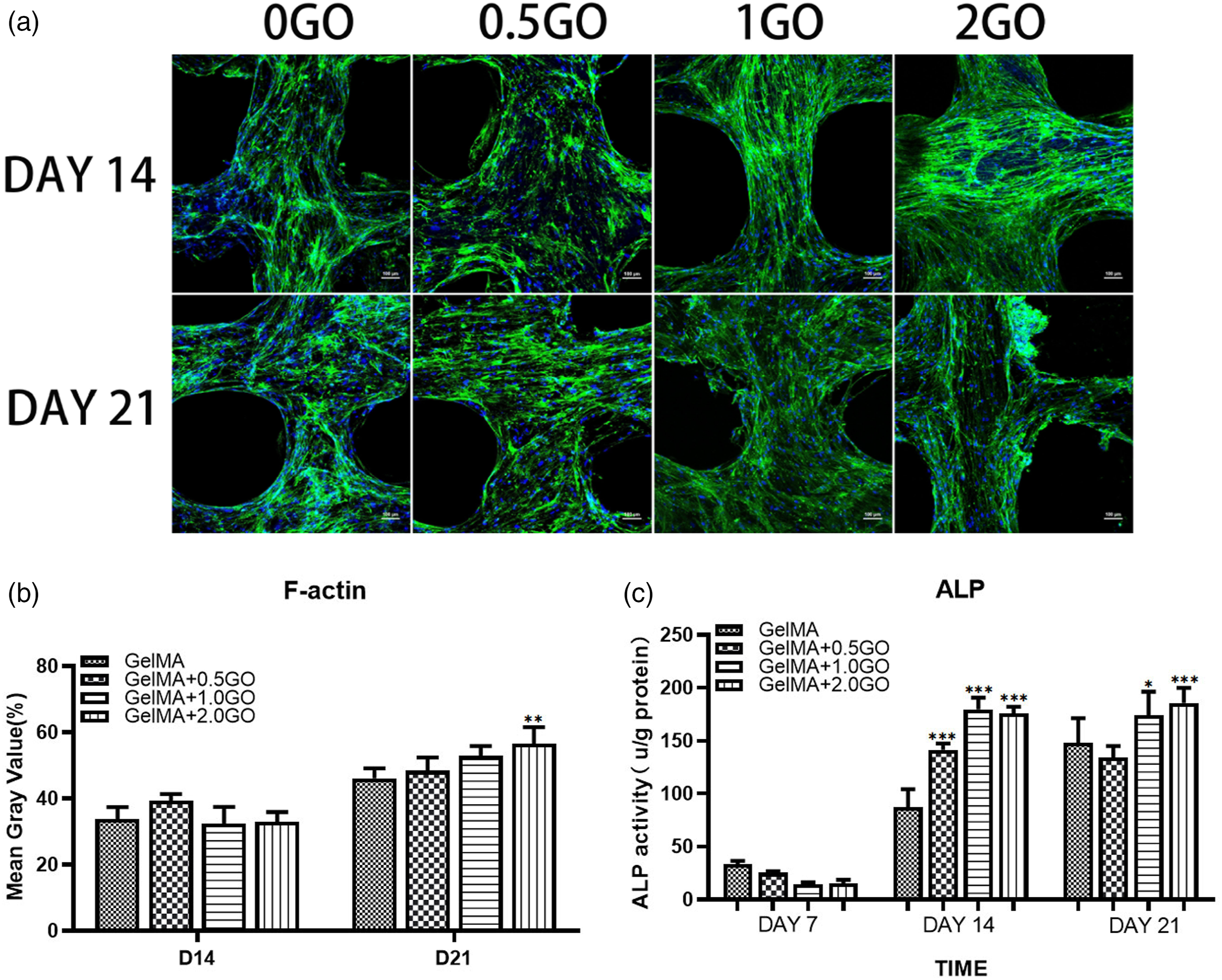
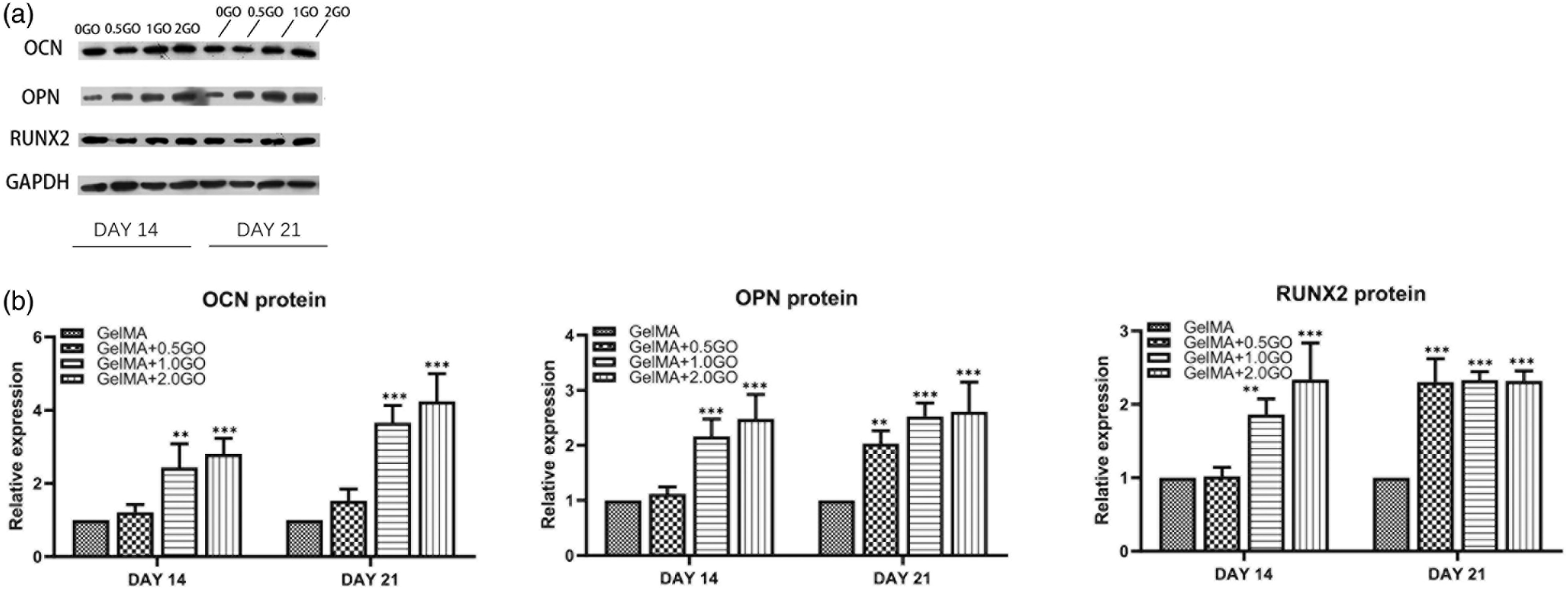
Finally, the expression of osteogenesis-related genes and proteins on the 3D bioprinted constructs was evaluated (Figures 5 and 6). After 2 weeks, a significant increase in OCN, OPN, and RUNX2 expression was observed in the GO group compared to that in the GelMA alone group. Moreover, after 3 weeks, the expression of OPN, RUNX2, and OCN was elevated in all groups, being significantly higher in the 2GO group than in the 0.5GO group. The expression trend of RUNX2 was the same as that at 2 weeks (Figure 5). Analysis of the respective encoded proteins at 2 weeks further confirmed that OCN levels were significantly higher in 1GO and 2GO groups than in the 0.5GO group; observed gene expression in the 0.5GO group was not reflected in protein expression. The trend on day 21 was similar to that observed on day 14. The levels of OPN and RUNX2 were significantly increased in the 1GO and 2GO groups after 14 days, and were elevated in the 0.5GO group after 21 days (Figure 6). Expression of osteogenesis-related genes in the 3D bioprinted structures. Expression of OCN, OPN, and RUNX2 at ( Expression of osteogenesis-related proteins in the 3D bioprinted structures. (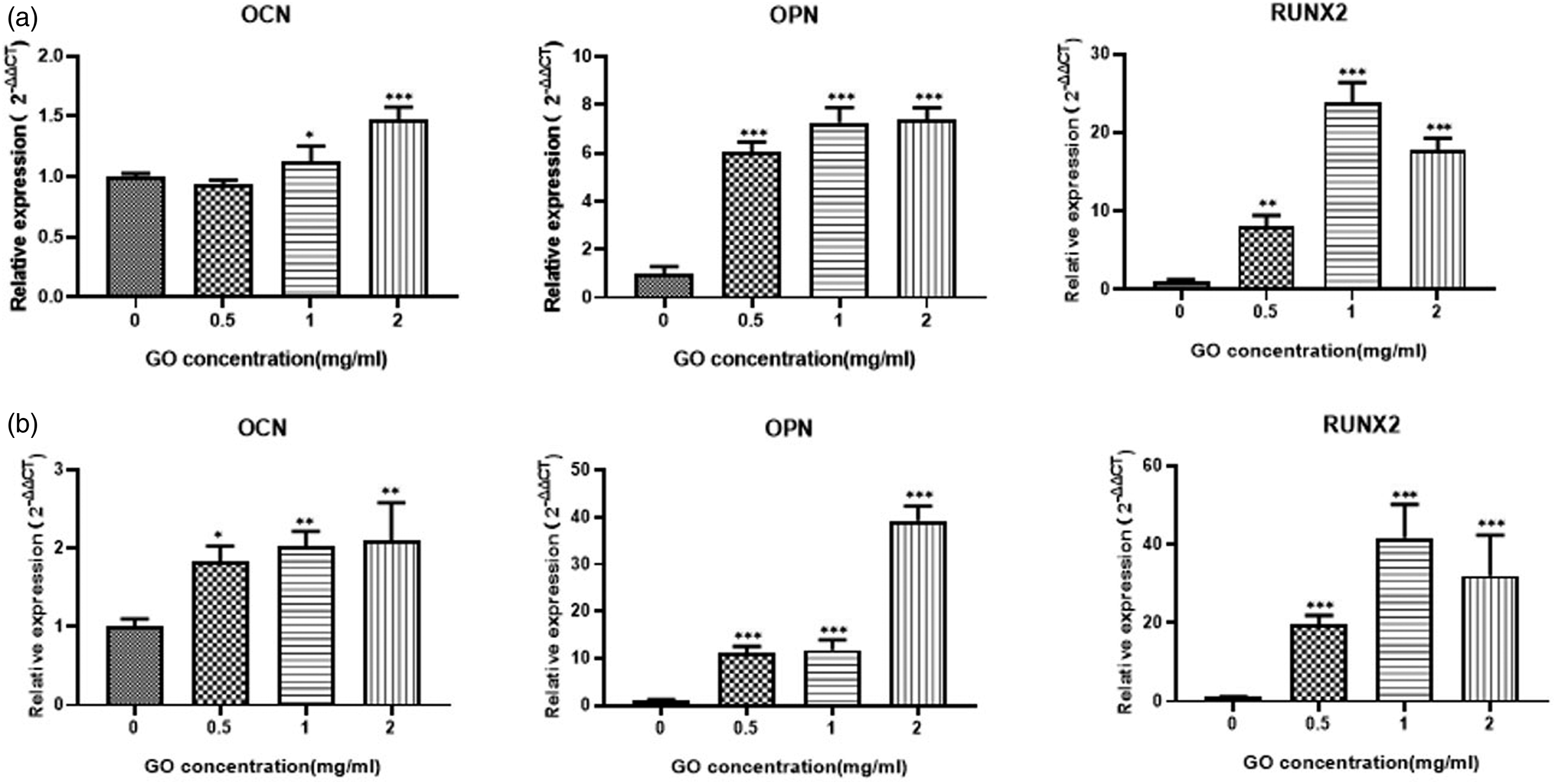
Discussion
The data collected in this study showed that the GelMA/GO complex bioink can promote osteogenic differentiation, with GO playing a driving role in the osteogenic differentiation process of MSCs. Choe et al. 21 had suggested that the effects of GO are related to the adsorption of osteogenic induction molecules by graphene, and had experimentally shown that scaffolds containing GO can adsorb more dexamethasone, which is an important induction molecule present in the osteogenic induction medium. Choe et al.’s data were similar to those of a previous study. 18
To investigate whether the addition of GO affects cell-material interactions and cell activities, the overall outcome of MSCs on days 1, 7, 14, and 21 after 3D bioprinting was evaluated. The cytotoxicity and ability of GO to promote cell proliferation were determined by comparing the cells cultured in GelMA scaffold to those cultured in GelMA/GO constructs. The concentration, size, and shape of GO can affect its cytotoxicity. Production of reactive oxygen species (ROS) is considered to be a frequent mechanism of GO toxicity; in particular, cell mortality was reported to be related to ROS production in or near cells. 13 Moreover, an increase in GO concentration has been suggested to lead to the formation of system viscosity and agglomerates, which limit GO dispersion and saturate the active sites in the system. 25 Herein, no significant decrease in cell activity was observed in presence of 2 mg/mL GO (the highest concentration tested). MSCs proliferated more rapidly in the 1GO and 2GO groups than in the rest, suggesting that the addition of appropriate concentrations of GO promotes cell proliferation. In addition, no difference in the final cell activity was observed between the 1GO and 2GO groups, which may be related to the saturation of the active site of the system.
Cell viability in the samples was examined on days 1, 7, 14, and 21 of culture. The high percentage of viable cells in the cultures demonstrated that the GelMA/GO composite has good biocompatibility for cell growth. Indeed, the cells showed continuous growth for over 21 days. Results after this time point indicated that the GO group had larger and more widely distributed cells, along with increased cell projection area, than those cultured in the GelMA scaffold. Hence, the results indicated that GO increases cell-cell interactions.
After 14 days of culture, ALP activity was significantly higher in the 1GO and 2GO groups than in the 0GO group. Nonetheless, after 21 days of culture, ALP activity in the 0GO group increased over that at 14 days, indicating that more cells were maintained in the osteogenic progenitor stage. Thereafter, ALP activity decreased in the 1GO and 2GO groups. As ALP plays a key role in early osteogenesis, with the hydrolysis of various phosphates promoting cell maturation and calcification, its upregulation represents a prerequisite for bone mineralization and subsequent maturation; therefore, decreases in ALP levels in the 1GO and 2GO groups may be related to the completion of the early calcification to late mineralization periods of the cells.
F-Actin is involved in various cellular functions, and its dynamics of aggregation and depolymerization are closely associated with cell morphology, with stronger F-actin backbones representing a morphological manifestation of osteogenic differentiation. 18 In this study, F-actin staining demonstrated the cell adhesion capacity of cells in the 2GO group to be increased, with extended actin spindles distributed layer by layer. Therefore, the enhanced intercellular interactions observed in the composite hydrogels further demonstrated the advantage of GO in providing better internal conditions for adhesion and proliferation within hydrogels.
qRT-PCR and western blot experiments were performed to further evaluate the effects of GO on the molecular landscape of osteogenic differentiation of ADSCs. After osteogenic induction, the expression of OPN, OCN, and RUNX2 was higher in the scaffolds of GO groups than in those of the 0GO group, except for RUNX2 levels in the 0.5GO group. OPN is widely distributed in bone tissue and can enhance the mechanical strength of the extracellular matrix by binding to type 1 collagen and OCN. 19 After 14 days of culture, the levels of OPN in the GO group were observed to be significantly higher than those in the GelMA group, whereas no significant difference was observed in the gene expression patterns between the 1GO and 2GO groups. On day 21, OPN expression increased in all samples, with the 2GO group showing a sustained increase in OPN expression, which was significantly higher than that in the other groups. However, the change in mRNA expression did not cause a change in the final OPN protein levels. OCN is a non-collagenous protein synthesized by osteoblasts during bone matrix mineralization, and represents an important marker of osteogenic activity and bone transformation. 26 OCN expression was elevated in the 1GO and 2GO groups at 2 weeks, although the 0.5GO group showed a statistically significant difference only at 3 weeks. Moreover, the trends of OCN protein levels were similar to those of its mRNA expression, with the 2GO group showing the highest levels of OCN, indicating a more pronounced stimulation of osteogenesis at this GO concentration. As a key and essential transcription factor in the osteogenic differentiation of MSCs, RUNX2 can initiate the transcription of osteogenic-related genes and promote protein expression; thus, it can be used as an indicator of osteoblast differentiation. High expression of RUNX2 can upregulate the expression of osteogenic-related factors, such as OCN, enhance ALP activity, promote the synthesis of extracellular matrix components, such as type I collagen, and promote mineralization.25,27 Herein, RUNX2 expression in the GO group was found to be significantly higher than that in the pure GelMA group at 14 days, and was gradually enhanced over time, with the GO group maintaining a clear advantage throughout the experimental duration. Regarding RUNX2 protein levels, no significant difference was observed between the groups at 2 weeks. Nonetheless, the protein levels increased in the 1GO and 2GO groups at 3 weeks, although only the change noted in 2GO was statistically significant. RUNX2 protein levels in the 0.5GO group never showed significant changes. Altogether, the results indicated that the effect of GO on RUNX2 is different from that on other osteogenic-related factors, although additional studies are warranted to validate the findings.
The present study showed that osteogenic capacity increased with increasing GO concentration; however, whether the increase in osteogenic capacity can be sustained with increasing GO remains to be answered. Considering that high concentrations of GO are cytotoxic, we considered that there would be a certain inflection point in the trend, which would still require further investigation. In this study, we constructed an inducible microenvironment by adding GO to the constructed particles, and GO at a concentration of 2 mg/mL had no significant toxic effect on the cells. However, we need to explore the relationship between this concentration and the induction effect in subsequent experiments. Additionally, owing to the condition, we were unable to conduct rheological testing to demonstrate the effect of GO addition on the mechanical properties, viscosity, shear dilution performance, and shear recovery ability of the bioink, which need to be uncovered in a subsequent study.
Conclusions
To develop an osteoinductive microenvironment for bone regeneration, we formulated GelMA/graphene oxide composite hydrogels and explored their feasibility in direct extrusion bioprinting. We demonstrated the feasibility of the strategy by detecting cell viability (>95%) in the cells printed within GelMA/GO scaffolds. Importantly, scaffolds with high graphene oxide content could stimulate osteogenic differentiation, which was evident by ALP activity assay, RT-qPCR, western blot, and immunostaining. From the data, GO was considered capable of promoting osteogenic differentiation, that of MSCs being stronger with increases in the concentration of GO over a certain range. Although the osteogenic capacity of the scaffold continued to increase with the increase in GO concentration, the increased content of GO may lead to unavoidable damage in the cell membrane during extrusion from the nozzle; the relationship between granular diameter and cell number will be discussed in the future.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the 2018 Project of Beijing Municipal Science and Technology Commission [grant number Z181100001718187].