
Abstract
The aim of this study was to investigate the potential of natural melanin nanoparticles (MNPs) as carriers for oxaliplatin (OXP) delivery, with glycol chitosan (GC) surface modification to improve dispersion behavior and drug interaction. The isolated MNPs were spherical with an average diameter of 148 ± 28 nm, which increased to 261 ± 39 nm after GC coating. The particle size decreased to 148 ± 59 nm after OXP loading. Physicochemical characterization confirmed surface modification and drug association. The OXP-loaded GC-MNP formulation showed a loading efficiency of 67.8 ± 4.6% and a biphasic release profile, with an initial rapid release followed by complete drug release within 24 h. Release kinetic analysis showed the best fit with the first-order model, indicating concentration-dependent release behavior. Biological studies demonstrated time-dependent responses in MCF-7 cells. All tested formulations showed low cytotoxicity at 6 h. However, OXP-loaded GC-MNP exhibited higher cytotoxicity than free OXP under selected conditions. Furthermore, concentration-dependent genotoxic effects were observed in comet assay experiments. These findings suggest that GC-coated melanin nanoparticles may serve as a promising carrier platform for short-term modulation of OXP delivery in breast cancer applications.
Introduction
Cancer is the second leading cause of mortality worldwide, following cardiovascular diseases, and is anticipated to become the foremost cause of death by 2030. 1 Due to its widespread occurrence, the development of effective treatments for cancer is of paramount importance. Despite the substantial advancement in surgical techniques, chemotherapy, and radiotherapy, cancer remains a primary cause of death and morbidity worldwide. 2 In most cases, surgical intervention is feasible in the early stages of the disease. Despite its effectiveness, radiation therapy carries risks to nearby healthy tissues, often results in adverse effects for patients. Similarly, chemotherapy, another cornerstone of cancer treatment, targets cancer cells, but can cause damage to healthy tissues, leading to hair loss, bone marrow suppression, reproductive system impairment, and gastrointestinal disturbances.3–8 Among the various types of cancer, one of the most common is breast cancer in women worldwide. Several platinum-based chemotherapy drugs, including carboplatin, cisplatin, and OXP, are routinely used in breast cancer treatment, with OXP exhibiting a relatively favorable side effect profile.9–11 Nevertheless, OXP continues to fail to fully mitigate side effects or ensure optimal therapeutic outcomes, highlighting the limitations of conventional cancer treatments.
Nanoparticles, with their remarkable drug bioavailability and potential for targeted delivery, represent a promising and innovative alternative to traditional cancer therapies. Especially, surface modified nanoparticles can acquire desirable properties, such as prolonged half-life, reduced toxicity, prevention of drug resistance, and enhanced accumulation at disease sites.12–16 With the use of nanoparticles as drug carriers, it is possible to prolong the half-life of drugs, enhance their solubility, and facilitate the controlled release of drugs.17,18 Due to their inherent biodegradability, biocompatibility, antioxidant properties, and widespread availability in nature, MNPs are a particularly promising candidate for drug delivery applications. 19 Further, surface modifications and combinations with other substances that enhance their functionality allow MNPs to be used for a variety of biomedical purposes. 20 Innovative nanoparticle surface coatings that address conflicting requirements can enable new applications to be developed. A nanoparticle’s surface charge and hydrophobicity are crucial factors in its design. Nanoparticles with a positive charge tend to adsorb on membranes and undergo a greater level of internalization compared to particles with a negative charge or neutral charge. A wide range of biomolecules interact with nanoparticles in vivo, and non-specific adsorptions may alter their physicochemical characteristics. 21 Previously, studies have examined polyvinyl alcohol and glycol chitosan complexes with MNP.22,23 As such, recent research has highlighted the potential of positively charged glycol chitosan for coating negatively charged melanin nanoparticles. 24 Cell viability assays revealed that cisplatin-loaded nanocarriers had significantly higher IC50 values than free CDDP, effectively meeting low dosage requirements while consistently eradicating liver cancer cells. This indicates their potential as a controlled release system in nanomedicine development. 24
The present study demonstrates that using MNPs is an effective method for delivering OXP due to various advantages, including the ability to accommodate hydrophobic drugs, outstanding biocompatibility, complete biodegradability, and enhanced stability compared to alternative nanoparticulate systems.25–29 Additionally, we previously demonstrated that GC modification of nanoparticle surfaces prevented the accumulation of melanin nanoparticles. 24 Thus, we postulate that OXP-loaded GC-modified MNPs could serve as efficacious carriers for delivering OXP, thereby potentially enhancing drug efficacy, refining pharmacokinetic properties, and enhancing anticancer efficacy while mitigating systemic toxicity. A comprehensive physicochemical analysis of the newly developed and optimized oxaliplatin nanoformulations was conducted as well as an assessment of their performance in vitro using cell culture models in terms of cell viability and genotoxicity.
Material and methods
Materials
Squid (Sepia officinalis) ink was purchased from Nortindal Sea Products Ltd (Spain). Glycol chitosan (GC), phosphate buffer saline (PBS, pH: 7.4), sodium chloride (NaCI), potassium chloride (KCI), potassium phosphate monobasic (KH2PO4) and sodium phosphate monobasic (NaH2PO4) were obtained from Sigma Aldrich. Oxaliplatin (OXP, 5 mg/mL) was purchased from Deva İlaç (Türkiye).
Preparation of GC-MNP and OXP-GC-MNP
MNPs extraction and purification from squid ink
The MNPs extraction and purification process from squid ink was performed according to the methods of Eom et al. (2017). 30 The squid ink was diluted five times with distilled water. Then, ink solutions were centrifuged at 10.000 rpm for 20 min. At the end of the centrifugation, supernatants were removed, and the remaining pellets were washed with distilled water 3 times to remove salts and impurities. Then, the collected pellets were dried in an oven at 50°C for 2 days.
Preparation of GC-MNP and OXP-GC-MNP
To prepare GC-MNP, 0.03 g glycol chitosan (GC) was dissolved in 5 mL of 1% (v/v) acetic acid solution and stirred at room temperature for 5 h to ensure complete dissolution. Separately, a stock melanin nanoparticle (MNP) suspension at 1% (w/v) was prepared by gravimetrically weighing dried MNP powder and dispersing it in deionized water to a final defined volume. Equal volumes (1:1, v/v) of the GC and MNP solutions were then combined and stirred overnight at 50 rpm at 37°C to promote uniform coating of MNPs with GC. The resulting mixture was centrifuged at 10,000 rpm for 20 min, and the supernatant was discarded. The dark-brown, gel-like extract obtained as pellet was collected and stored at 4°C for further analysis.
To conjugate oxaliplatin (OXP) to GC-MNP, 0.25 mg of OXP powder was dissolved in 500 µL of phosphate-buffered saline (PBS, pH 7.4). This OXP solution was added to the GC-MNP suspension and incubated under gentle shaking for 24 h at room temperature to allow for non-covalent drug loading. The mixture was then centrifuged at 10,000 rpm for 20 min to separate unbound OXP. The supernatant was collected and analyzed using a UV–Vis spectrophotometer at 277 nm to determine the concentration of unbound drug. Drug loading efficiency (%) was calculated using equation (1):
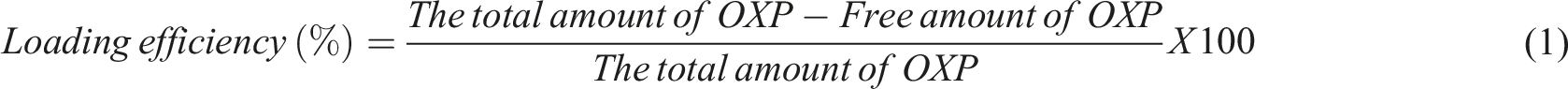
The final OXP-GC-MNP product appeared as a dark-brown, gel-like dry extract, stored for further in vitro studies.
MNP, GC-MNP and OXP-GC-MNP characterization
SEM (FEI Quanta 400F, Hillsboro, OR, USA) was used to characterize the morphology of MNP, OXP-MNP, GC-MNP, and OXP-GC-MNP. A gold-palladium (Au-Pd) mixture was sputtered onto the samples before they were examined, and the SEM images were taken at a voltage of 7.5 kV. ImageJ® software (NIH, MD, USA) was used to determine the average nanoparticle diameters for each formulation. All data was presented as the mean ± SD (n = 100).
The chemical structure of GC, GC-MNPs, OXP-GC-MNPs and compatibility between components were evaluated with the Attenuated Total Reflectance Fourier Transform Infrared Spectrum (ATR-FTIR) device. The FTIR spectrometer (Perkin Elmer Spectrum Two, Waltham, MH, USA) was performed with a collected spectrum in the range of 4000-1000 cm−1.
The thermal properties and compatibility of the components within the nanocomplexes were assessed through thermogravimetric analysis (TGA, TA Instruments Co., DE, USA). Each sample was placed in aluminum pans and subjected to heating from 30 to 600°C at a rate of 10°C/min in an inert nitrogen atmosphere.
In vitro drug release
Investigation of OXP release from MNP and GC-MNP was carried out in vitro using a phosphate-buffered saline (PBS, pH 7.4). The formulations of MNP, GC-MNPs, and OXP-GC-MNPs were suspended in PBS solution (pH 7.4, 1 mg/mL) and incubated at 37°C with stirring at 300 rpm. Samples were collected at specified intervals and centrifuged at 10,000 rpm for 20 min to separate the released OXP from the nanocomplexes. The concentration of OXP in the supernatants was monitored at 277 nm using a UV-VIS spectrophotometer.
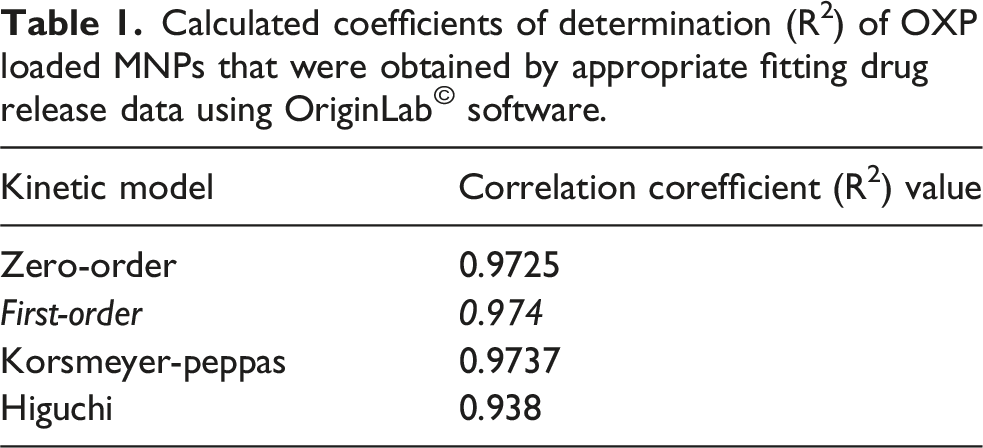
Based on the release profiles of OXP from GC-MNPs, four kinetic models have been examined: zero-order, first-order, simplified Higuchi, and Korsmeyer-Peppas. Several independent measurements were conducted to obtain the data from the nanoformulations. The data were presented as mean values with standard deviations. The 95% confidence intervals for the differences in the mean dissolution profiles at each dissolution time point were calculated.
In vitro cell culture studies
Cytotoxicity
The breast cancer cell line MCF-7 (ATCC, HTB-22) was used for the cytotoxicity tests. Before cell seeding, cells were maintained with Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 1% penicillin-streptomycin, 10% fetal bovine serum (FBS), and humidified atmosphere containing 5% CO2 at 37°C to grow to confluency (≅80%). Cells were then harvested with 0.25% trypsin-EDTA, seeded in a 96-well plate at a concentration of 1.3 × 105 per well, and incubated for 24 h at 37°C and 5% CO2. Following, the culture medium was replaced with fresh serum-free medium containing various concentrations (1-100 µg/ml) of OXP, GC-MNP, and OXP-GC-MNP. The untreated cells served as the control group, and they were incubated at 37°C with 5% CO2 for 6, 12, 18, and 24 h. After the incubation period, the medium was discarded from the wells and replaced with 80 µL of serum-free media and 20 µL (5 mg/mL) of the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution into each well. The plate was incubated for 3 h (37°C, 5% CO2) to generate formazan crystals. Following incubation, the MTT solution was carefully removed from all wells, and 100 µL of DMSO was added to each well to dissolve the resulting formazan crystals. The plate was then placed in a shaking incubator for 30 min to ensure complete solubilization. The intensity of the purple color was measured at 560 nm using a plate reader (BioTek, Synergy Neo 2). Cell viability was calculated by accepting the viability of the control group as 100%.
Genotoxicity
The alkaline single cell gel electrophoresis (Comet) method was used to determine the genotoxic effects of OXP, GC-MNP, and OXP-GC-MNP in MCF-7 cells. After the cells were grown in 25 cm2 flasks and trypsinized, they were seeded into 6-well plates at the concentration of 3 × 104 cells/well to be treated with the solutions of nanocomplexes. Plates were incubated for 24-h at 37°C, containing 5% CO2 atmosphere, and then exposed to cytotoxic concentrations of OXP and obtained structures in serum-free DMEM (25, 50, and 100 µg/mL) for 6 h. The 6-h exposure was chosen because it was the period during which the highest cytotoxicity was observed. Serum-free DMEM was used as the negative control. Following exposure, cells were trypsinized and washed with PBS for one time. 50 µL of cell-PBS suspension was mixed with 100 µL of LMA (1%) and then spread on slides coated with NMA (1%). After the slides were kept at 4°C for the solidification of agar, the cells were kept in lysis solution (2.5 M NaCl, 100 mM EDTA, 100 mM Tris, 1% sodium sarcosinate, pH 10.0 with Triton-X-100, and 10% DMSO) at 4°C for 1 h. At the end of this period, the slides were placed in the electrophoresis tank and kept in the electrophoresis solution (1 mM sodium EDTA and 300 mM NaOH, pH 13.0) for 20 min. The electrophoresis was then performed at 4°C using 24 V and adjusting the current to 300 mA by raising or lowering the buffer level. For neutralization, the slides were kept in PBS solution for 15 min. After the neutralization step, the slides were treated with 50%, 75%, and 98% alcohol, respectively. DNA damage in cells was assessed by staining the slides with EtBr and then analyzing 100 cells on each slide using Comet Analysis Software, version 4.0 (Kinetic Imaging Ltd, Liverpool, UK). Results were given as tail intensity (%). 31
Statistical analysis
The results are expressed as the mean ± standard deviation (SD) of at least three replicates. Two-Way ANOVA tests were followed by Tukey’s and Games-Howell’s multiple comparison post-tests to evaluate statistical significance. Statistical analysis was performed using Excel and GraphPad Prism 10 software (GraphPad) based on *p < 0.05, **p < 0.01 and ***p < 0.001 for significant differences.
Results and discussion
Characterizations of nanoparticles
One of the major concerns in the delivery of drug loaded nanoparticles to their target sites is their size, due to its impact on biodistribution, cellular uptake, and clearance rates from the body, all of which can significantly affect the therapeutic efficacy and safety profile of the treatment.32,33 According to SEM analysis, MNPs exhibited a spherical morphology with a solid, rough texture and measured approximately 148 nm ± 28 nm (n = 100) in diameter [Figure 1(a1)]. These particles showed low aggregation and relatively uniform distribution, as reflected in the narrow size distribution histogram [Figure 1(a2)]. OXP loading to MNPs did not cause a notable change in either average particle size or agglomeration behavior, resulting in a measured diameter of 133 nm ± 19 nm (n = 100) [Figure 1(c1,2)]. However, the size of GC-MNP increased after GC coating, likely due to structural collapse during dehydration, and was measured as 261 nm ± 39 nm (n = 20) [Figure 1(b1,2)]. It should be noted that a lower particle count was analyzed for GC-MNPs because the hydrogel-like network formed upon glycol chitosan modification led to partial aggregation, reducing the number of well-dispersed and individually measurable nanoparticles under SEM. As observed in the SEM images, visible aggregation and surface irregularities were more apparent in this group, suggesting the presence of a soft outer coating layer. After the washing process and OXP loading, the size of nanoparticles was found to be 148 nm ± 59 nm (n = 100), exhibiting moderate aggregation yet a still distinguishable spherical morphology [Figure 1(d1,2)]. The body’s filtration systems, such as the kidneys, eliminate nanoparticles below 10 nm, while those larger than 200 nm are removed by phagocytic cells in the reticuloendothelial system (RES).
14
Several studies have demonstrated that nanoparticles in the 20–200 nm range accumulate more efficiently in tumor tissues, as they evade RES clearance and renal filtration.34–36 Thus, both OXP-loaded MNPs, before and after GC coating, fall within the optimal size range for enhanced tumor targeting, aligning with the design criteria for effective nanocarrier-based delivery systems. SEM images of (a1) MNPs, (b1) GC-MNP, (c1) OXP-MNP, and (d1) OXP-GC-MNP, and corresponding particle size distribution histograms (a2, b2, c2, d2). Size distribution was calculated using n = 100 particles for MNPs, OXP-MNP, and OXP-GC-MNP, and n = 20 for GC-MNP. SEM images were captured at 80,000× (a1–b1) and 100,000× (c1–d1) magnifications (scale bar = 1 μm). OriginLab© software was used for data evaluation and statistical analyses.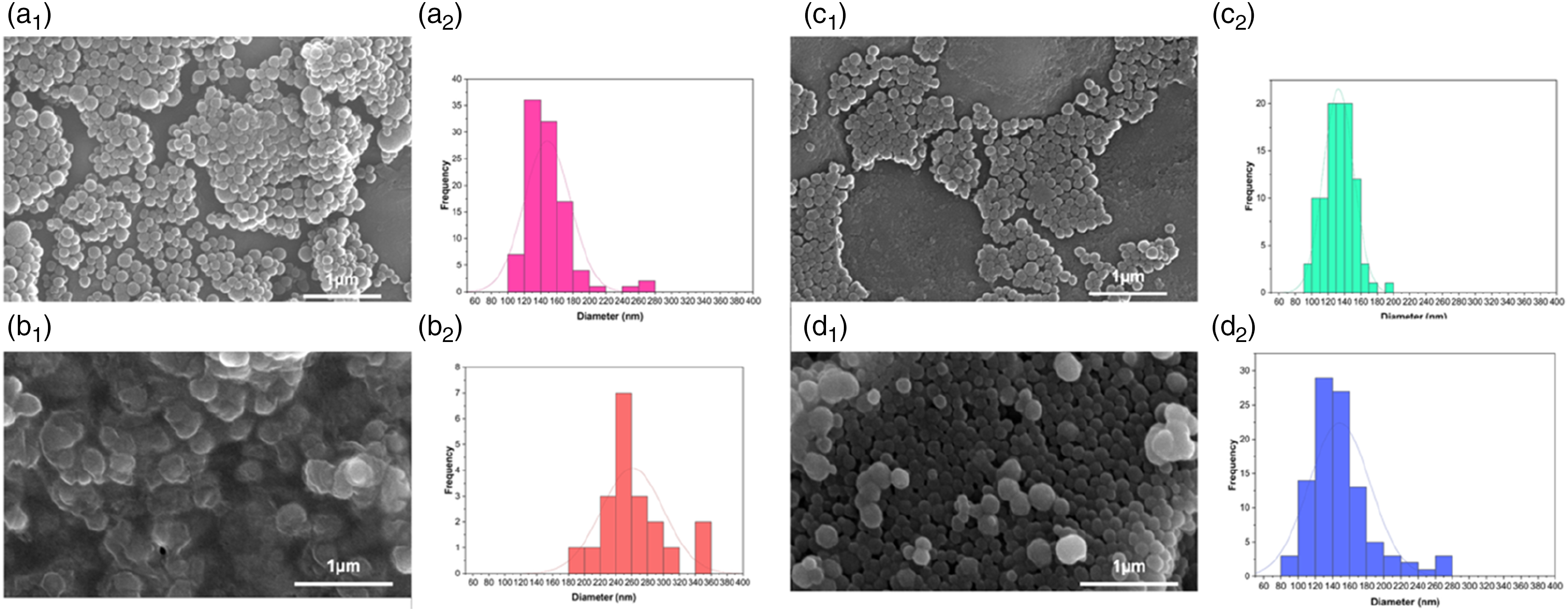
The ATR-FTIR spectra of GC, GC-MNP, and OXP-GC-MNP are presented in Figure 2(a). In the spectrum of GC, bands at 3355 cm‒1 and 3289 cm‒1 correspond to O–H and N–H stretching vibrations. Characteristic amide and carboxyl group peaks were observed at 1755 cm‒1 (C=O stretching), 1645 cm‒1 (amide I), 1568 cm‒1 (amide II), 1140 cm‒1 (C–O–C stretching), and 1070 cm‒1 (C–O stretching).
37
The GC-MNP spectrum retained these characteristic features, with a notable shift of the amide I band from 1645 cm‒1 to 1635 cm‒1, likely arising from hydrogen bonding or secondary interactions between GC functional groups and melanin moieties such as dihydroxyindole and indolequinone [Figure 2(b)].24,26,38 (a) ATR-FTIR spectra of GC, GC-MNP, and OXP-GC-MNP powders; (b) normalized and magnified view of the C=O stretching region (∼1720 cm‒1); (c) normalized and magnified view of the amide I region (∼1600 cm‒1), indicating interactions between GC and MNPs following OXP loading.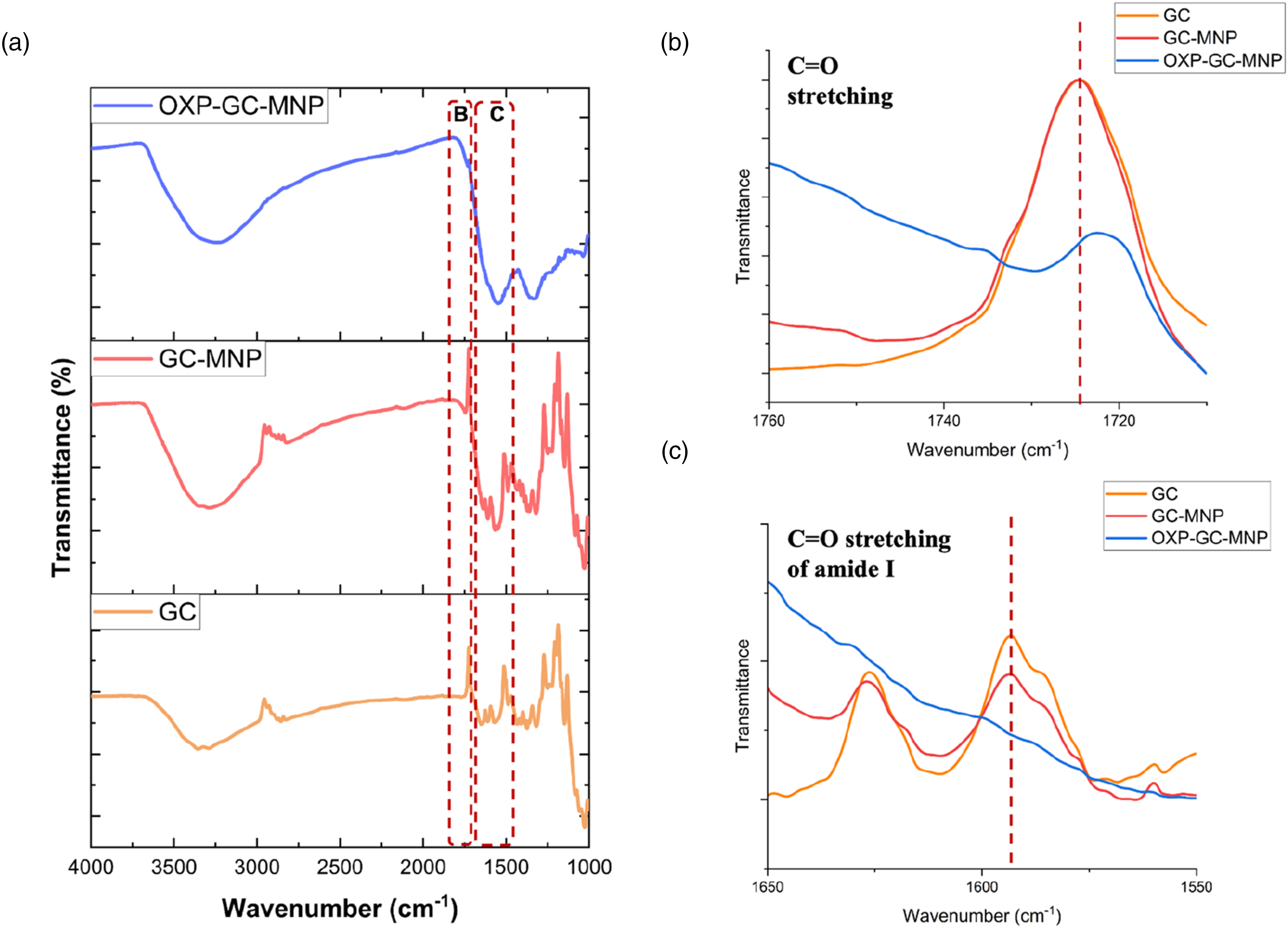
For the OXP-GC-MNP formulation, several distinct spectral changes were observed. In addition to the O–H/N–H and C–O stretching bands remaining visible at 3233 cm‒1 and 1088 cm‒1, OXP loading induced clear changes in carbonyl-associated regions. The C=O stretching peak shifted from 1745 cm‒1 (GC-MNP) to 1730 cm‒1, and the amide I band shifted from 1635 cm‒1 to 1600 cm‒1 [Figure 2(c)]. The observed band shifts indicated the molecular interactions of OXP with the GC-modified melanin nanoparticles confirming the successful association of OXP with the OXP-GC-MNP structure.
The thermal decomposition behaviors of MNPs, OXP, GC, GC-MNPs, and OXP-GC-MNPs were analyzed using thermogravimetric analysis (TGA), as illustrated in Figure 3. The results indicated that the GC coating process resulted in a shift to higher temperatures, thereby delaying thermal degradation. Conversely, the loading of OXP with GC-MNPs significantly altered thermal stability in the initial degradation stage. However, the thermal behavior during the second stage of degradation remained unchanged. Thermogravimetric spectra of OXP, GC, MNPs, GC-MNPs, and OXP-GC-MNPs powders.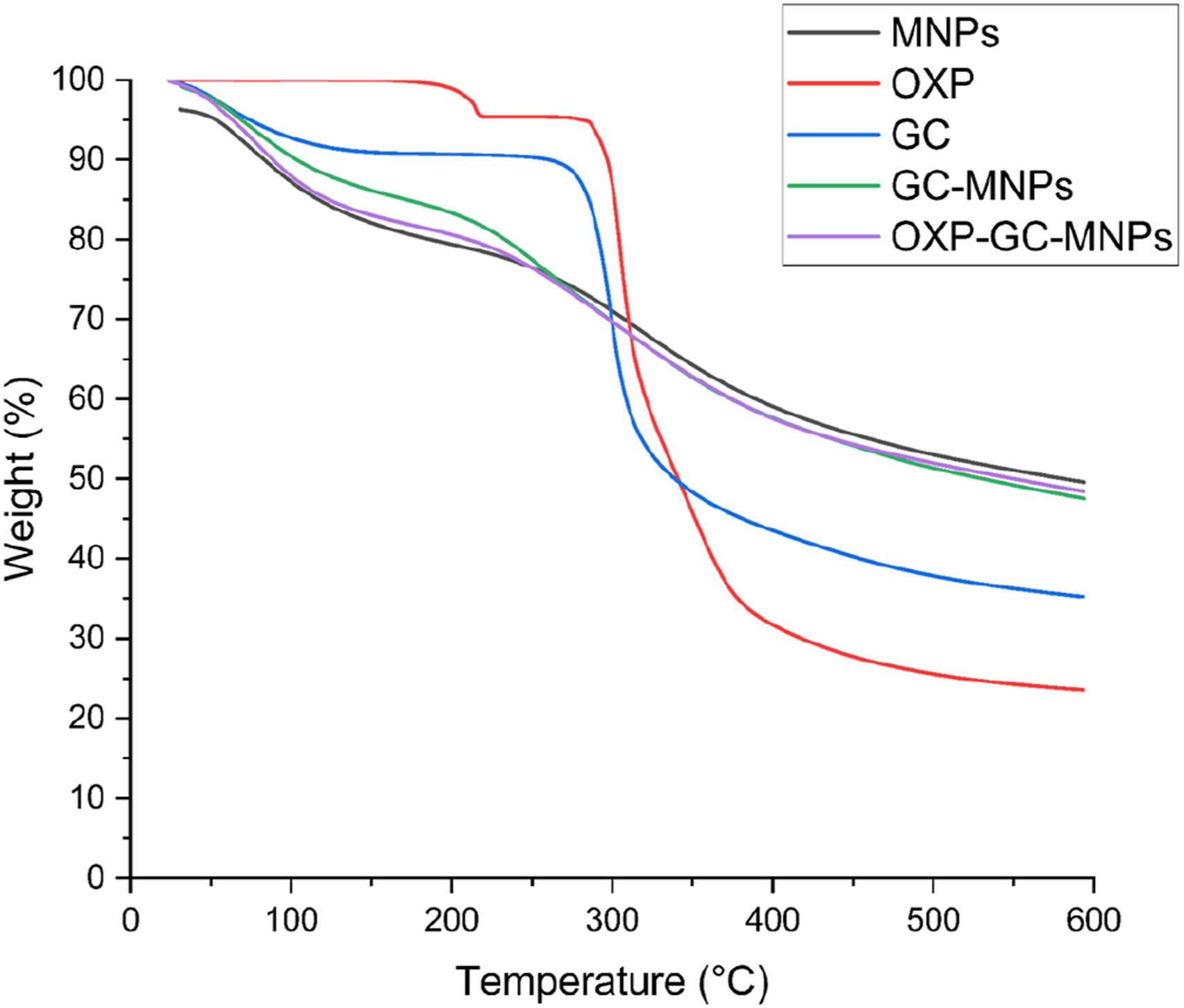
In vitro drug release
The drug loading efficiency calculated using equation (1) based on an initial OXP amount of 0.25 mg was 67.8 ± 4.6%. This value is in accordance with previously reported oxaliplatin carrier systems such as hydrogels, superparamagnetic iron oxide nanoparticles, chitosan nanoparticles, and PEGylated half generation polyamidoamine (PAMAM) dendrimers with loading efficiencies around 69.90% ± 0.88 %, 55.2% ± 4.8%, 91.4%, and 75.69% ± 8.90 %, respectively.39–42
The OXP-loaded GC-MNP exhibited a biphasic release profile with approximately 80% of the drug released in the first 10 h and complete release within 24 h (Figure 4). The first burst phase can be attributed to the surface associated OXP molecules that are more easily desorbed into the release medium.
43
The slower phase that followed suggests ongoing release of the more strongly associated fractions of the drug. Although this profile is not indicative of a long-term depot system, prolonging OXP availability over 24 h may still be preferable to rapid depletion of free oxaliplatin in biological systems.
44
Such short-term sustained exposure may increase local availability of the drug in the early therapeutic window. OXP release profiles from GC-coated MNPs.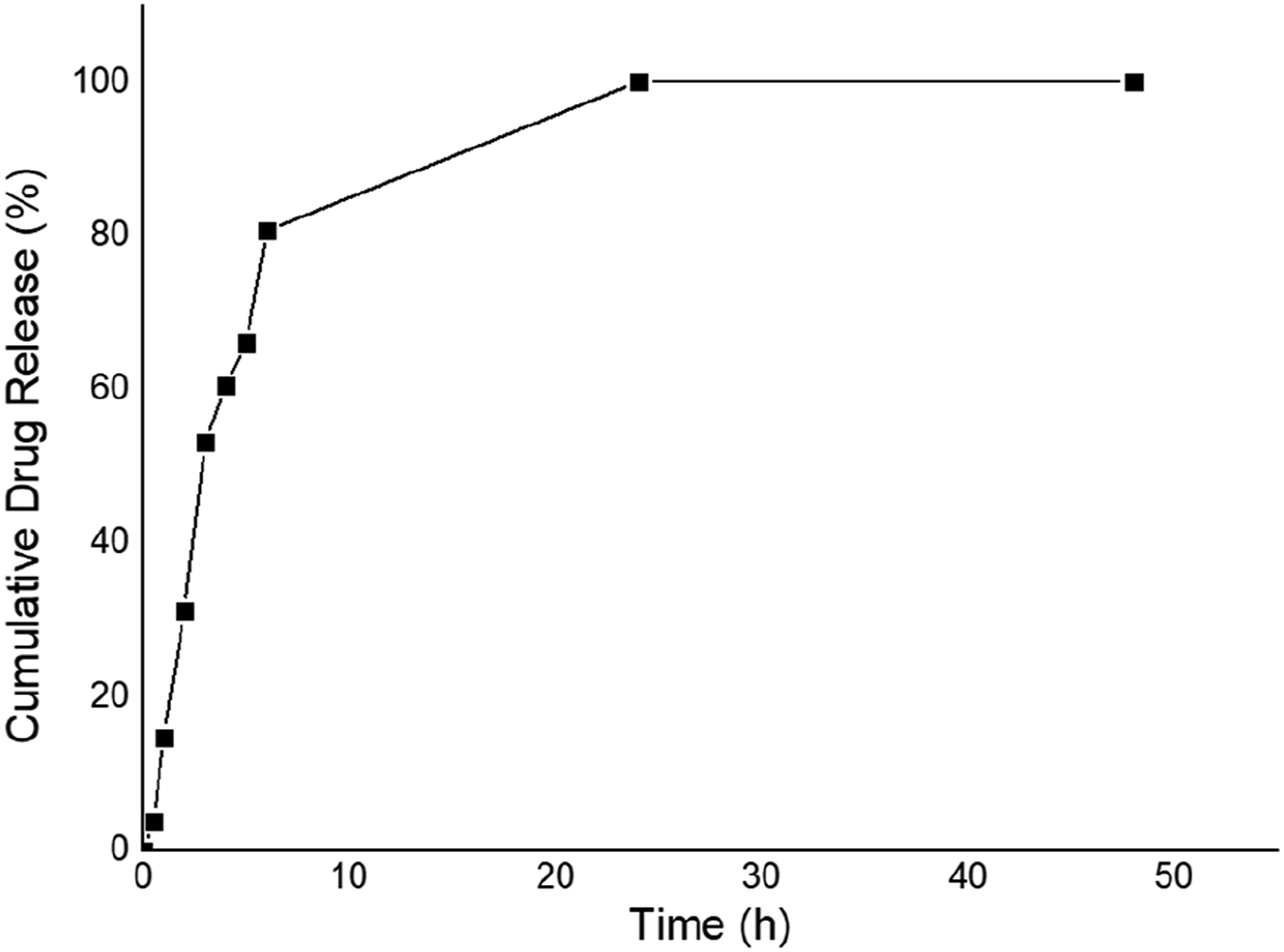
The release behavior of the present study was different from previously reported oxaliplatin carrier systems. Human serum albumin–aminated mesoporous silica nanoparticles showed a slow and incomplete release profile, releasing 53.37% of OXP at 120 h. 45 Solid lipid nanoparticles showed 99.39% cumulative release after 144 h which is more prolonged release duration. 46 In contrast, superparamagnetic metal-organic frameworks showed 85% release within 9 h and no further significant release up to 30 h. 47 Compared with these systems, OXP-loaded GC-MNP exhibited a faster biphasic release profile with ∼80% release within 10 h and complete release within 24 h. These differences suggest that the OXP release kinetics are strongly dependent on the carrier composition, surface interactions and structural properties. The present formulation may thus be more suitable for applications where rapid initial availability of the drug followed by short-term sustained exposure is desired rather than prolonged release over several days.
Calculated coefficients of determination (R2) of OXP loaded MNPs that were obtained by appropriate fitting drug release data using OriginLab© software.
In vitro cell culture studies
The MTT results obtained in this study revealed a time-dependent cytotoxicity profile that does not follow a classical dose–response relationship (Figure 5). In particular, the low level of cytotoxicity observed at the early time point (6 h) across all groups is consistent with the known mechanism of action of oxaliplatin (Figure 5(a)). Platinum-based agents exert their cytotoxic effects primarily through the formation of DNA crosslinks, a process that requires time to induce detectable cellular damage.
47
Therefore, limited toxicity at early time points is an expected outcome. Results of MTT experiments with MCF-7 cells to determine the cytotoxic effects of OXP, GC-MNP, and OXP-GC-MNP. The MTT assay was carried out after 6 (a)), 12 (b), 18 (c), and 24 (d) hours of incubation with OXP, GC-MNP, and OXP-GC-MNP with MCF-7. Data are presented as a percentage of cell survival (untreated cells were assumed to have 100% survival). The data represent the average viability values ±SD of three independent samples. Statistical significance between time points was determined using two-way ANOVA followed by post hoc analysis (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).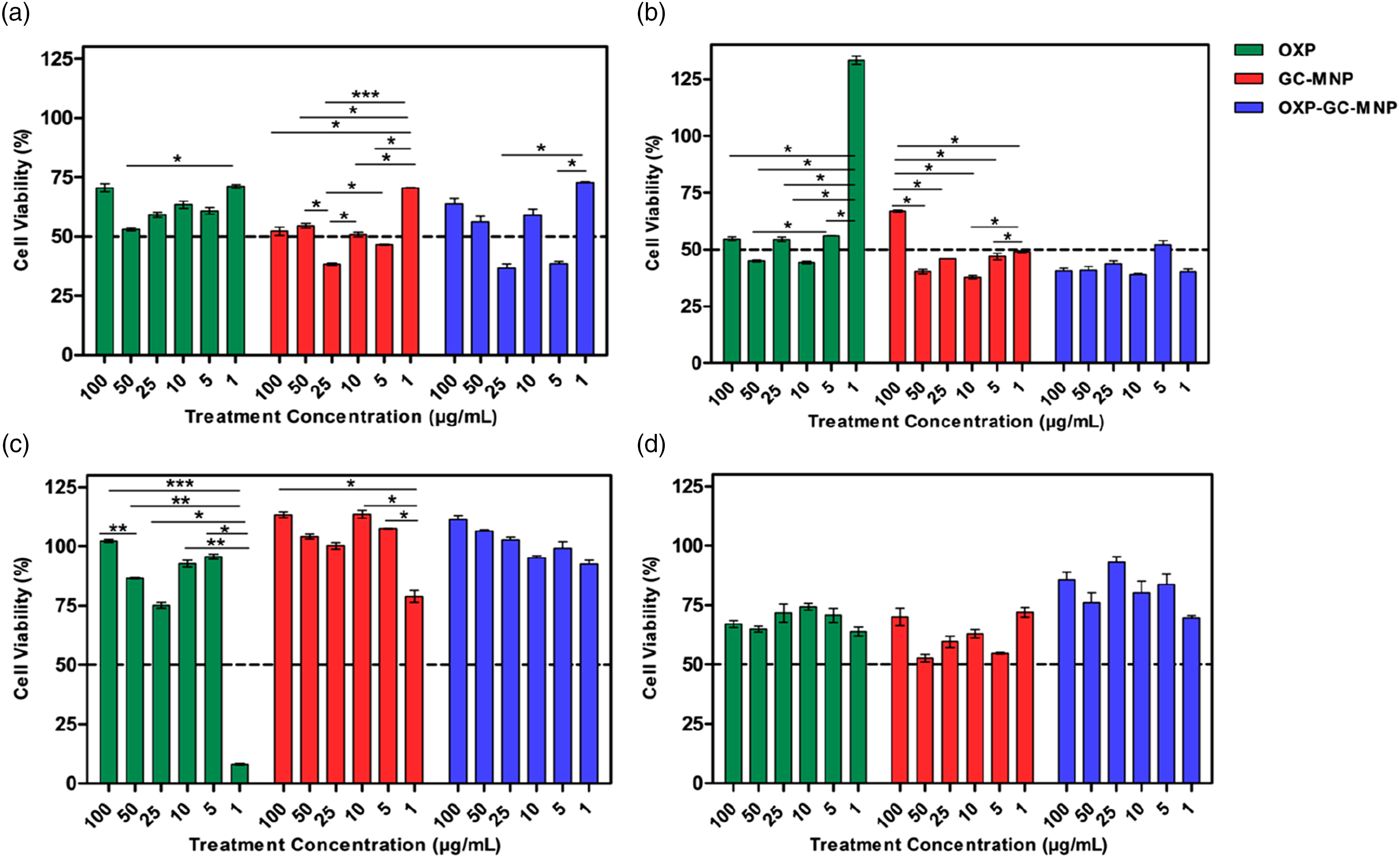
In contrast, the pronounced fluctuations observed at 12 and 18 h (Figure 5(b), and (c)), along with the absence of a clear dose–response relationship, suggest that the system suggests that additional formulation-related factors contribute by the drug effect and instead involves more complex mechanisms. One of the key contributing factors is likely the drug release kinetics of the nanoparticle system. In line with our findings, the initial burst release observed in nanoparticle systems can lead to a rapid increase in local oxaliplatin concentration within the cellular environment. This may enhance cytotoxicity at certain concentrations, whereas the subsequent slower release phase may result in reduced effects over time. Such biphasic release behavior has been widely reported in nanoparticle-based drug delivery systems.48,49
Furthermore, the role of glycol chitosan (GC) coating in modulating cellular interactions should also be considered. Due to its cationic nature, GC can enhance electrostatic interactions with the negatively charged cell membrane, thereby facilitating cellular uptake of nanoparticles.50,51 This enhanced uptake may increase cytotoxicity at certain time points due to more efficient intracellular drug delivery, while at other time points, controlled release behavior may lead to reduced cytotoxic effects. Therefore, the observation that GC exhibits both toxicity-enhancing and toxicity-reducing effects at different time points can be attributed to the dynamic nature of the system.
Another notable finding is the observation of cell viability values exceeding 100% in some groups (Figure 5(b), and (c)). This phenomenon has been frequently reported in the literature and is generally associated with changes in cellular metabolic activity rather than an actual increase in cell number. The MTT assay measures mitochondrial enzyme activity rather than direct cell count, 52 and thus increased metabolic activity can result in elevated MTT signals independent of cell proliferation. 53
In this context, the redox-active and antioxidant properties of the melanin nanoparticles used in this study may play a critical role. Melanin is known to act as a free radical scavenger and to participate in redox reactions.54,55 These properties may reduce intracellular oxidative stress and enhance cellular metabolic activity. Indeed, previous studies have demonstrated that melanin nanoparticles can increase cell viability, likely due to their antioxidant capacity.56,57 This effect may explain the elevated viability observed, particularly at the 24 h time point. (Figure 5(d)).
In addition, potential interference of melanin nanoparticles with the MTT assay should also be considered. Since the reduction of tetrazolium salts is a redox-dependent process, redox-active biomaterials such as melanin may directly influence the assay outcome.53,58 Such interactions may artificially increase the measured signal, leading to overestimation of cell viability beyond actual biological levels.
For all treatment groups (OXP, GC-MNP, and OXP–GC-MNP), statistically significant differences were observed between all time points (6, 12, 18, and 24 h) at all tested concentrations (p < 0.0001). Specifically, for each concentration, cell viability differed significantly across time points, indicating that the cytotoxic response varies over time even at the same dose. These findings demonstrate that cytotoxicity is strongly time-dependent under the experimental conditions.
Genotoxicity studies
When the genotoxic effects were evaluated as a result of 6-h exposure, where the structures showed the highest cytotoxic effect at cytotoxic concentrations, a significant increase in the genotoxic effect was observed with the increase in concentration. The fact that the obtained structures cause more genotoxic effects, especially when compared to OXP, suggests that they are promising in the treatment of breast cancer (Figure 6). This increase supports the fact that an increase in the genotoxic effect, which is one of the basic mechanisms of action of anticancer drugs, causes a decrease in cancer cell viability.
59
Genotoxic effects of OXP, GC-MNP, and OXP-GC-MNP on the MCF-7 cell line after 6 h of incubation. The data represent the average viability values ±SD of three independent samples. NC: Negative control (DMEM without serum) * shows statistically important difference with negative control (p < 0.05).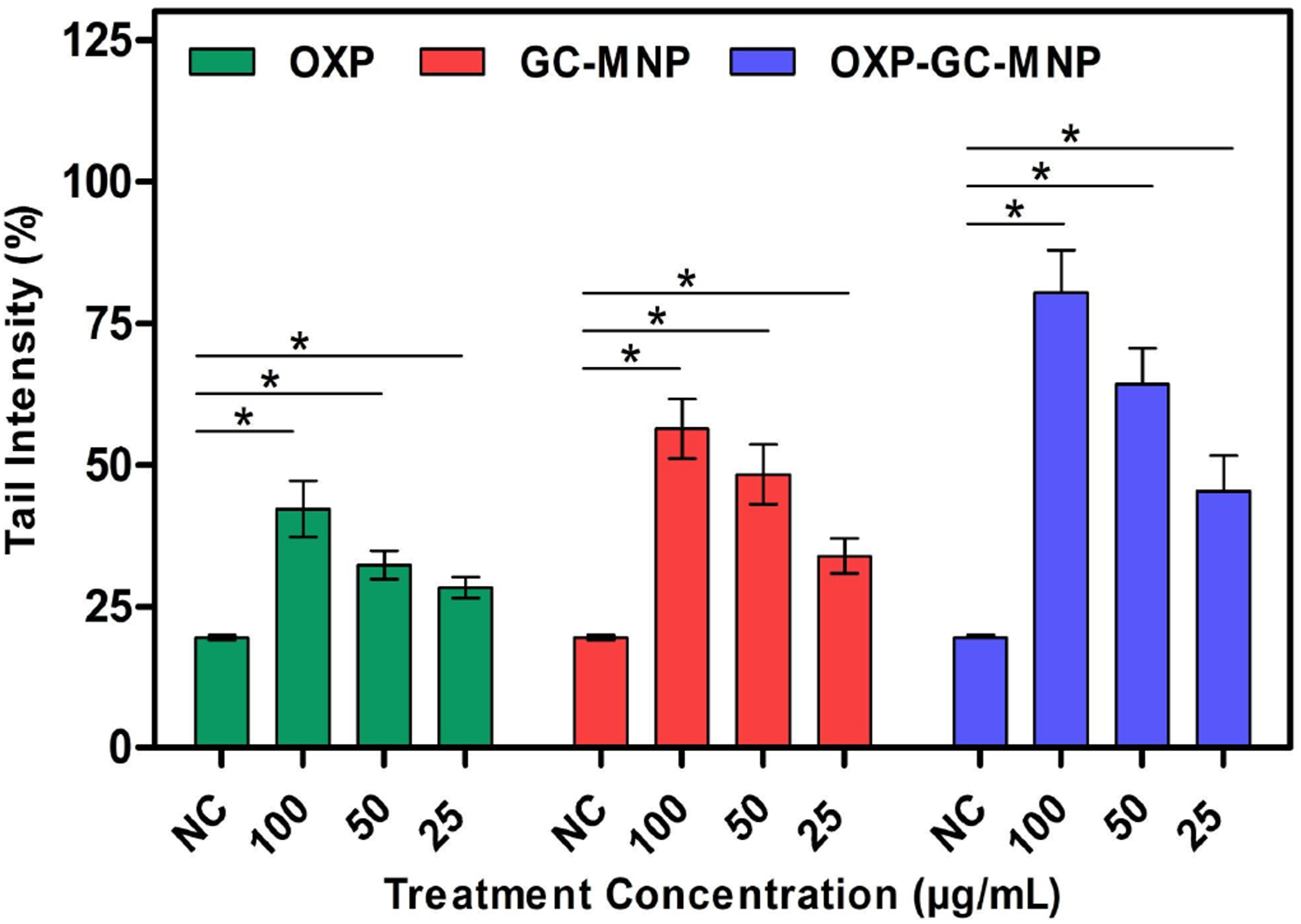
Although the nanoparticle formulations exhibited higher levels of DNA damage compared to free OXP, as indicated by the Comet assay, it is important to note that genotoxicity alone does not directly translate into therapeutic efficacy. The increased DNA damage observed in cancer cells should therefore be interpreted as part of a broader cellular response rather than as a standalone indicator of anticancer activity. In this context, the genotoxic effects were evaluated alongside reductions in cell viability, which together suggest an enhanced stress response in malignant cells.
Conclusion
Thus, a glycol chitosan-coated natural melanin nanoparticle platform was developed for oxaliplatin loading and evaluated as a potential drug delivery system for breast cancer applications. The formulation showed an OXP loading efficiency of 67.8 ± 4.6% together with characteristic physicochemical changes following surface modification and drug association. It also exhibited a biphasic release profile, with an initial rapid release followed by short-term sustained exposure over 24 h. Biological experiments demonstrated time-dependent effects in MCF-7 cells, suggesting that drug concentration, release kinetics, and nanoparticle–cell interactions collectively influence the overall response of the system. These findings indicate that GC-coated melanin nanoparticles may serve as a promising carrier platform for short-term modulation of OXP delivery.
This work represents in vitro proof-of-concept study. Future investigations will include additional cancer models, serum stability studies, targeting strategies, and in vivo pharmacokinetic evaluation. Moreover, theranostic applications may also be explored, given the inherent metal-binding and imaging potential of melanin-based materials.
Footnotes
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by Scientific and Technical Research Council of Türkiye (TÜBİTAK 2210-C) by providing Güleycan Dedecengiz Varol was a recipient of the TÜBİTAK 2210-C MSc Scholarship.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.