
Abstract
The cocirculation of CRF01_AE and CRF07_BC among men who have sex with men (MSM) in China may facilitate the emergence of genetically complex HIV-1 recombinants. Here, we identified and characterized a novel second-generation HIV-1 CRF01_AE/CRF07_BC recombinant, designated GY0192, from a 25-year-old MSM individual in Guiyang, Guizhou Province, southwest China, using near full-length genome (NFLG) analysis. Recombination analyses identified four breakpoints at HXB2 positions 3,125, 5,687, 6,375, and 9,176, generating a distinct five-segment mosaic genome. Subregion phylogenetic analyses showed that the two CRF01_AE-derived fragments clustered with the CRF01_AE cluster 5 lineage, while all three CRF07_BC-derived fragments grouped with CRF07_BC lineages frequently circulating among MSM in China, indicating that both parental components of GY0192 were phylogenetically related to lineages frequently reported among MSM populations in China. The virus was also predicted to be CCR5-tropic, adding epidemiological relevance because CCR5-tropic viruses are commonly involved in transmission and early infection. Together, these findings identify GY0192 as a distinct CRF01_AE/CRF07_BC mosaic and suggest that cocirculating MSM-associated HIV-1 lineages may provide opportunities for interlineage recombination. This case expands the known spectrum of CRF01_AE/CRF07_BC recombinants in Guizhou and underscores the value of NFLG-based surveillance for detecting underrecognized HIV-1 genetic complexity in southwest China.
Keywords
Introduction
HIV-1 is a highly diverse and rapidly evolving virus, and its extensive genetic diversity remains a major challenge for vaccine development. Inter-subtype recombination is a major driver of this genetic complexity, contributing to the generation of circulating recombinant forms (CRFs) and unique recombinant forms that continue to fuel viral evolution and epidemic spread. 1
In China, men who have sex with men (MSM) have become one of the key populations driving the contemporary HIV-1 epidemic. 2 Among MSM, CRF01_AE and CRF07_BC are two predominant lineages, and their sustained cocirculation likely provides repeated opportunities for superinfection and recombination. 3 The recent identification of multiple second-generation recombinants among Chinese MSM further underscores ongoing viral diversification in MSM-associated HIV-1 epidemics.2,4
Southwest China remains an important region for HIV-1 molecular evolution and recombinant emergence. 5 Although neighboring provinces such as Yunnan, Guangxi, and Sichuan have reported substantial viral diversity and frequent novel recombinants, recent reports from Guizhou have been relatively limited.4,6,7 This disparity may reflect insufficient genomic surveillance rather than a true absence of recombination, highlighting the need for intensified near full-length genome (NFLG)-based studies to better resolve local recombinant complexity. 8
Here, we identified and characterized, by NFLG analysis, a novel second-generation HIV-1 CRF01_AE/CRF07_BC recombinant, designated GY0192, from an MSM individual in Guiyang, Guizhou Province, China. Its distinct mosaic structure provides additional molecular evidence that cocirculation of major HIV-1 lineages associated with MSM populations in Guizhou may contribute to the emergence of new recombinant forms.
Material and Methods
In the current study, a plasma sample was collected in Guiyang City, Guizhou Province, southwest China, from an HIV-1-positive participant designated GY0192. GY0192 was a 25-year-old Han Chinese man, a university graduate, and a resident of Guiyang who self-reported sex with men. He was diagnosed as HIV-1 antibody-positive during a pre-employment physical examination in January 2025. At diagnosis, his baseline CD4+ T-cell count was 501 cells/µL. After enrollment in the national free antiretroviral treatment program, his CD4+ T-cell count increased to 585 cells/µL after 5 months and to 801 cells/µL after 11 months of treatment. This study was approved by the Ethics Committee of Guizhou Provincial People’s Hospital.
The methods used in the current study for amplification and sequencing of GY0192 were the same as those described earlier. 9 HIV-1 RNA was isolated from 200 µL of the patient’s plasma sample using the QIAamp Viral Mini Kit (Qiagen, Germany) following the manufacturer’s protocol. Nested PCR for NFLG amplification was performed using two sets of primers designed to amplify the 5′ and 3′ halves of the HIV-1 genome. PCR was conducted using serially diluted cDNA templates under the following cycling conditions for both rounds: initial denaturation at 94°C for 3 min; 35 cycles of 94°C for 20 s, 60°C for 30 s, and 68°C for 4 min; and a final extension at 68°C for 10 min. Positive PCR products were purified and sequenced using a QIAquick Gel Extraction Kit (Qiagen), and chromatograms were inspected, edited, and assembled using Sequencher v4.10.1.
HIV-1 reference NFLG sequences, including representative sequences of subtypes A1, A2, B, C, D, F1, F2, G, H, J, and K and CRFs CRF01_AE, CRF07_BC, and CRF08_BC, were downloaded from the Los Alamos National Laboratory HIV Sequence Database. To evaluate whether GY0192 represented a distinct recombinant pattern, available and published CRF01_AE/CRF07_BC recombinants were retrieved, and their breakpoint patterns and segmental compositions were compared with those of GY0192. Recombination signals, breakpoint positions, and segmental composition were identified and confirmed using the Recombinant Identification Program, SimPlot v3.5.1 similarity plot and Bootscan analyses, and jpHMM. Breakpoint positions were reported according to the HXB2 numbering system.
Phylogenetic analyses of the NFLG and each recombinant fragment were performed using FastTree under the generalized time-reversible model to generate approximately maximum-likelihood trees. To provide standard bootstrap validation, the topology and lineage assignment of the NFLG and subregion trees were further evaluated using the neighbor-joining method in Molecular Evolutionary Genetics Analysis (MEGA) with 1,000 bootstrap replicates. Bootstrap values ≥70% in the MEGA neighbor-joining analysis were considered to indicate phylogenetic support.
HIV-1 coreceptor usage was predicted based on the env V3 loop sequence using Geno2pheno[coreceptor], a V3 loop-based genotypic coreceptor tropism prediction tool, and WebPSSM, as described in previous studies. 10 For Geno2pheno[coreceptor], a false-positive rate (FPR) cutoff of 2.5% was used. FPR values below the selected cutoff were interpreted as supporting CXCR4-capable tropism, whereas FPR values at or above the cutoff were interpreted as not supporting CXCR4-capable tropism. WebPSSM was used as an additional V3 loop-based prediction tool, and the final tropism assignment was made based on the concordant genotypic prediction results from both tools.
Results
In this study, the NFLG sequence obtained from subject GY0192 spanned HXB2 positions 790–9,599, covering the HIV-1 coding region from gag to nef and extending into the 3′ long terminal repeat (LTR). Phylogenetic analysis based on the NFLG showed that GY0192 clustered with CRF01_AE reference sequences, but formed a distinct monophyletic branch that was relatively distant from other CRF01_AE strains (Fig. 1).
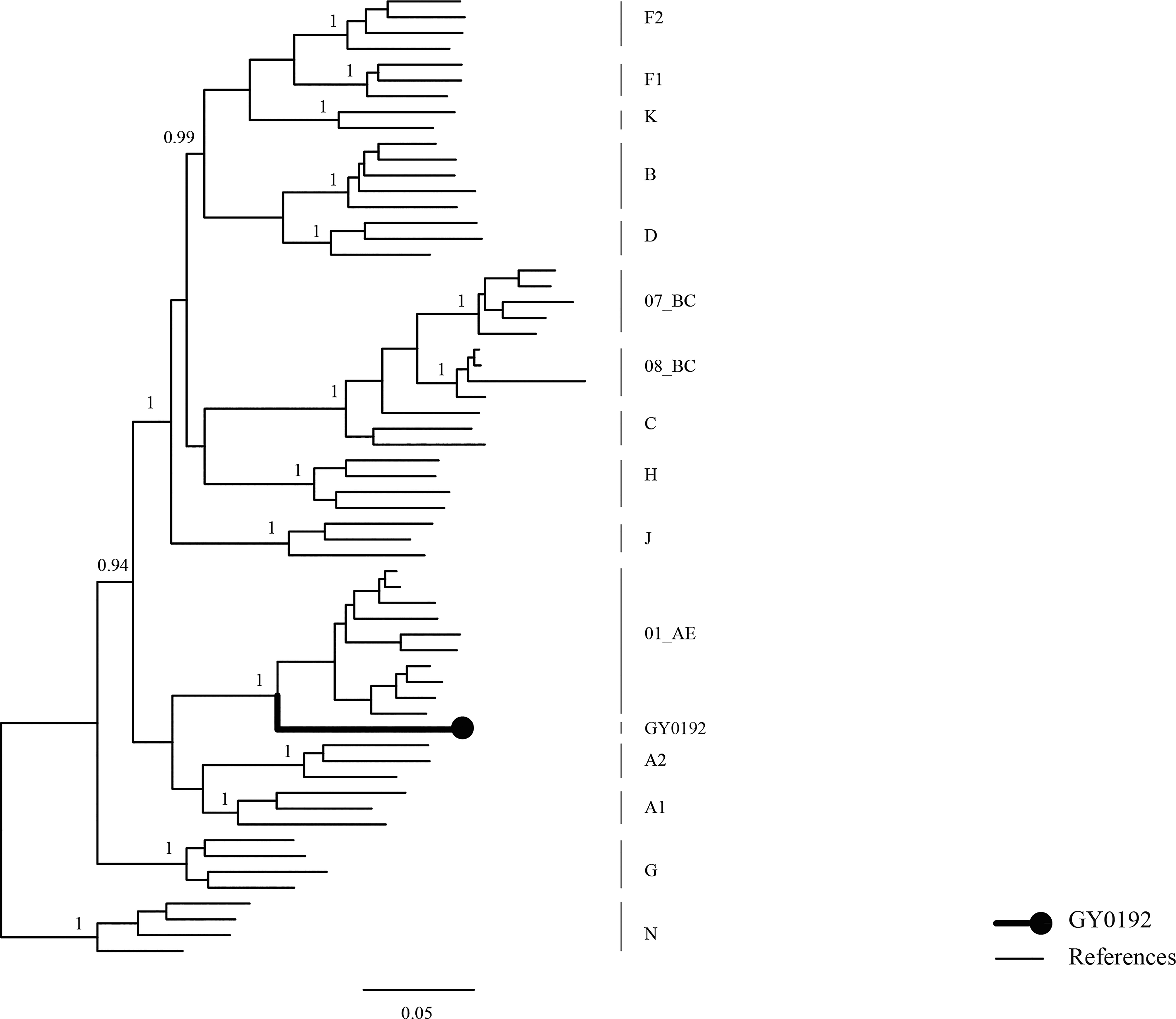
Phylogenetic analysis of the NFLG sequence of GY0192 and HIV-1 reference sequences. The displayed tree was generated using FastTree under the generalized time-reversible model as an approximately maximum-likelihood tree. The topology and lineage assignment were further validated using the neighbor-joining method in MEGA with 1,000 bootstrap replicates. HIV-1 group M subtypes, sub-subtypes, and selected CRFs were used as reference sequences. GY0192 is indicated by a bold black line and a solid circle. The scale bar represents 5% genetic distance. CRFs, circulating recombinant forms; NFLG, near full-length genome.
Recombination analyses using jpHMM and SimPlot showed that the GY0192 genome was composed of CRF01_AE and CRF07_BC, with multiple CRF07_BC fragments inserted into a CRF01_AE backbone (Fig. 2A). Bootscanning analysis identified four recombination breakpoints at HXB2 positions 3,125, 5,687, 6,375, and 9,176, located within the pol, vpr, env, and nef/3′ LTR regions, respectively. Accordingly, the genome of GY0192 was divided into five mosaic fragments: fragment I, CRF07_BC (HXB2: 790–3,125); fragment II, CRF01_AE (HXB2: 3,126–5,687); fragment III, CRF07_BC (HXB2: 5,688–6,375); fragment IV, CRF01_AE (HXB2: 6,376–9,176); and fragment V, CRF07_BC (HXB2: 9,177–9,599) (Fig. 2C). Importantly, comparison with available and published CRF01_AE/CRF07_BC recombinants showed that the breakpoint pattern and segmental composition of GY0192 were distinct from those of previously reported recombinants. These findings support the classification of GY0192 as a distinct novel recombinant form.
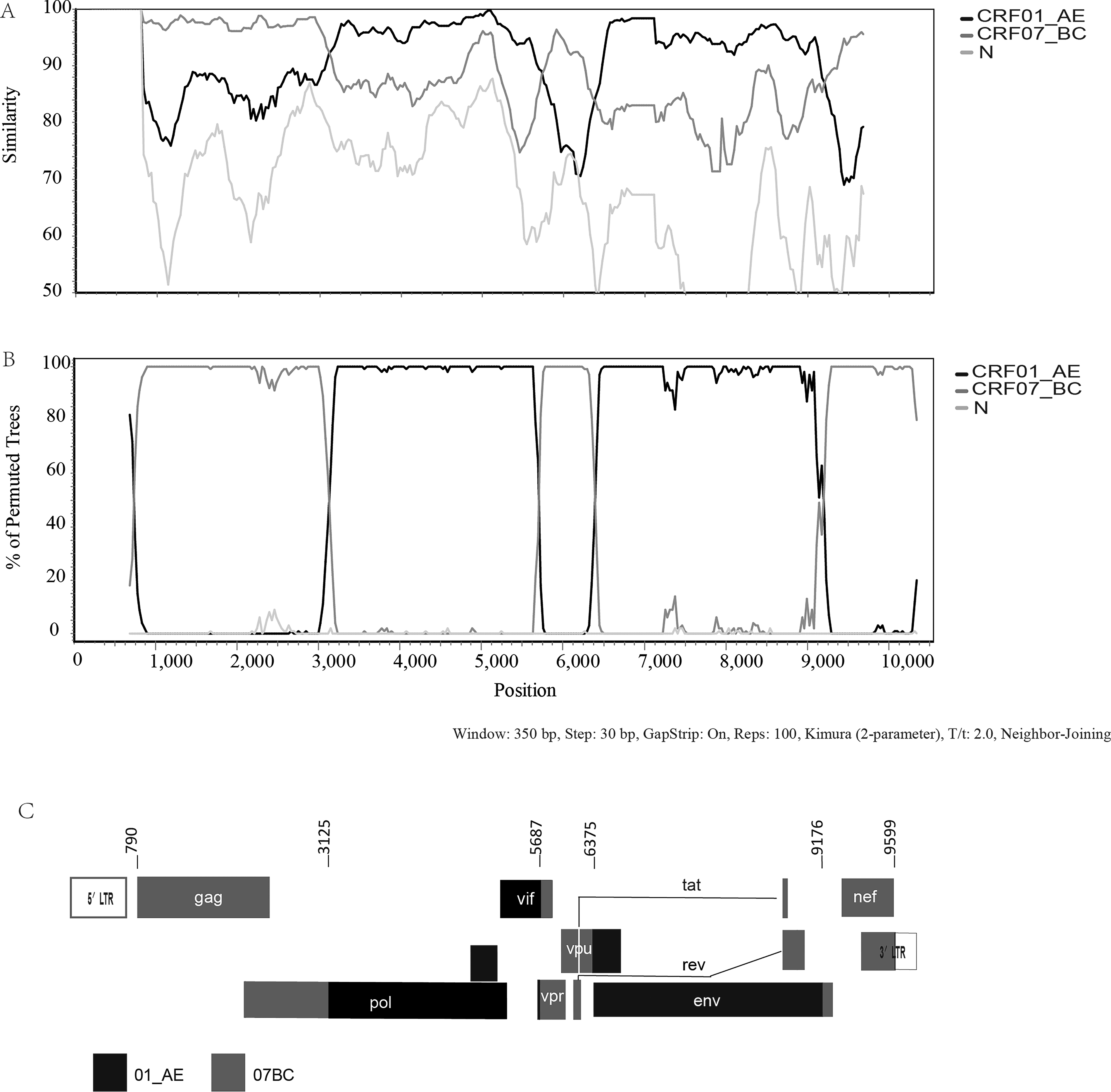
Recombination analyses of the novel second-generation recombinant GY0192.
Further subgenomic phylogenetic analyses revealed that recombinant regions II and IV clustered with the CRF01_AE cluster 5 lineage, one of the major lineages circulating among MSM in China 11 (Fig. 3). Notably, all three CRF07_BC-derived fragments (regions I, III, and V) also grouped with CRF07_BC lineages frequently identified in MSM populations in China. Taken together, these findings indicate that both the CRF01_AE-derived and CRF07_BC-derived components of GY0192 were phylogenetically related to lineages frequently reported among MSM populations in China. In addition, V3 loop-based genotypic prediction using Geno2pheno[coreceptor] and WebPSSM indicated that GY0192 was CCR5-tropic. Geno2pheno[coreceptor] yielded an FPR of 35.1%, which was above the selected 2.5% cutoff, and WebPSSM predicted an R5 phenotype.
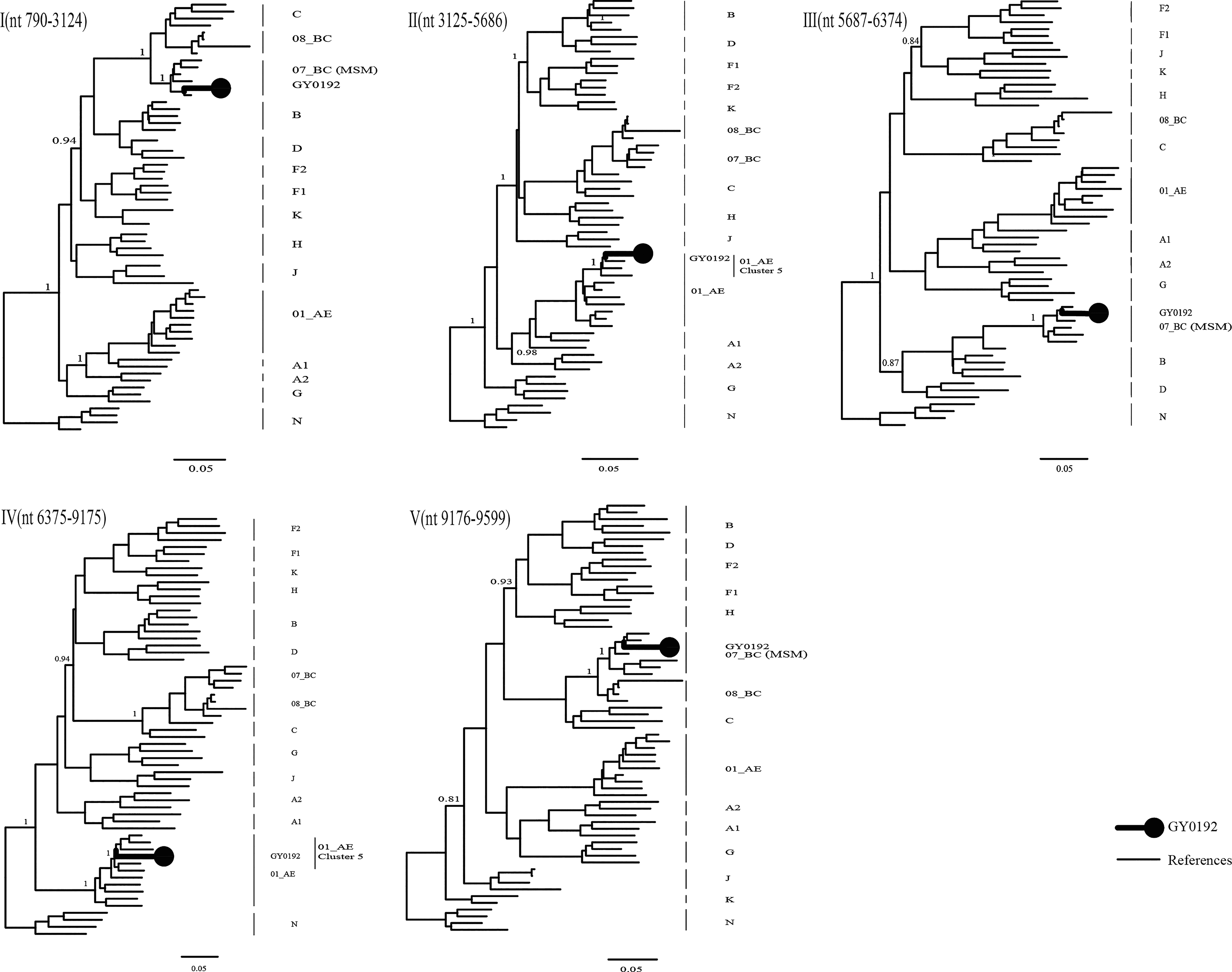
Phylogenetic analysis of the five recombinant fragments of GY0192 defined by Bootscan analysis. The displayed trees were generated using FastTree under the generalized time-reversible model as approximately maximum-likelihood trees. The topology and parental origin of each fragment were further validated using the neighbor-joining method in MEGA with 1,000 bootstrap replicates. GY0192 is indicated by a bold black line and a solid circle, and HIV-1 reference sequences are labeled on the right side of each tree. The scale bars represent 0.05 substitutions per site.
Discussion
As HIV-1 infections among MSM in China have increased rapidly, the genetic diversity of circulating viruses has also expanded. In recent years, multiple second-generation recombinant forms composed of CRF01_AE and CRF07_BC have been reported in different provinces, particularly among MSM populations. For example, three novel CRF01_AE/CRF07_BC recombinants were identified in Hebei Province, 12 two additional recombinants (BDD034A and BDL060) were reported among MSM in the same region, 13 and the second-generation recombinant CRF135_0107 was subsequently characterized in China. 14 Together, these findings suggest that the cocirculation of CRF01_AE and CRF07_BC in MSM populations may provide opportunities for ongoing recombination and viral diversification.
Notably, the breakpoint pattern and segmental composition of GY0192 were distinct from previously reported CRF01_AE/CRF07_BC recombinants, including those identified in Guizhou.15,16 The NFLG of GY0192 exhibited a relatively complex five-segment mosaic structure composed of alternating CRF07_BC- and CRF01_AE-derived fragments. More importantly, subgenomic phylogenetic analyses showed that the CRF01_AE-derived regions clustered with the CRF01_AE cluster 5 lineage, while all three CRF07_BC-derived fragments grouped with CRF07_BC lineages frequently circulating among MSM in China. These findings indicate that both parental components of GY0192 were related to lineages frequently observed among MSM populations in China. Therefore, GY0192 may have emerged in the context of cocirculation and recombination of major HIV-1 lineages associated with MSM populations, although direct evidence for a strictly local transmission origin is not available from this single-sequence study.
Guizhou occupies an epidemiologically important position in southwest China. It borders Yunnan and Guangxi, two provinces with substantial HIV burdens, marked genetic diversity, and active molecular transmission networks. 17 In the context of increasing population mobility and ongoing regional strain exchange in southwest China, this geographic setting may facilitate cross-regional viral introduction, local cocirculation of major HIV-1 lineages, and onward dissemination. 17 Recent evidence from HIV-1-infected MSM in Guiyang showed that CRF07_BC and CRF01_AE were commonly detected in this population and that molecular transmission network analysis can identify clusters with potential transmission risk. 18 Together, these factors may create favorable conditions for sustained cocirculation of distinct HIV-1 lineages and, consequently, for repeated recombination. Against this background, the identification of GY0192 provides further molecular evidence that the cocirculation of HIV-1 lineages associated with MSM populations may provide opportunities for the emergence of genetically complex recombinant forms in Guizhou.
Another noteworthy finding is that V3 loop-based genotypic prediction suggested CCR5 tropism for GY0192. Although this inference was based on genotypic prediction rather than phenotypic entry assays, it remains epidemiologically relevant because CCR5-tropic viruses are more commonly associated with transmission and early-stage infection. This feature provides an additional clue supporting the public health relevance of continued molecular surveillance for newly arising recombinant strains.
An additional point of epidemiological interest is that GY0192 was identified in a 25-year-old MSM individual. Although this study is based on a single case, the detection of a genetically complex second-generation recombinant in a young adult suggests that younger sexually active MSM populations may represent an important setting for ongoing viral spread and diversification. This observation supports the need for continued emphasis on early diagnosis, timely linkage to care, and targeted prevention in younger at-risk populations.
In conclusion, GY0192 represents a distinct novel second-generation CRF01_AE/CRF07_BC recombinant identified in an MSM individual in Guizhou, and its mosaic structure suggests that cocirculation of major HIV-1 lineages associated with MSM populations may contribute to continued viral diversification in this region. The phylogenetic affinity of both parental components to MSM-associated lineages suggests that GY0192 may have arisen in a context where these lineages overlap, enabling interlineage recombination. Broader population-level sampling is needed to clarify its transmission history and geographic origin. These findings underscore the value of NFLG-based molecular surveillance for uncovering underrecognized HIV-1 genetic complexity and improving molecular epidemiological monitoring in southwest China.
Sequence Data
The NFLG sequence of GY0192 reported in this study has been deposited in GenBank under accession number PZ291593.
Authors' Contributions
Z.G., D.L., and H.Z., contributed equally to this work. Z.G., D.L., and H.Z., performed the experiments, analyzed the data, and drafted the manuscript. Z.N., H.Z., and Q.L., contributed to sample collection, data curation, and methodological support. J.Z., conceived and supervised the study, provided critical revision of the manuscript, and approved the final version for submission.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by the National Natural Science Foundation of China (Regional Fund, Grant No. 82260658); the Guizhou Provincial Science and Technology Program (Qian Ke He Foundation–ZK [2023] General 215); the Guizhou Province High-Level Innovative Talent Program (gzwjrs2023-002); the Guizhou Provincial Key Technology R&D Program No. [2021] normal 057; and the Guizhou Province Returned Overseas Scholars Innovation and Entrepreneurship Excellence Funding Program (Contract No. [2025] 012).