
Abstract
The long-term co-circulation of various HIV-1 subtypes and circulating recombinant forms (CRFs) in China has created an environment conducive to viral recombination. This study identified a novel HIV-1 CRF resulting from the recombination of CRF07_BC and CRF55_01B in Xishuangbanna Prefecture, Yunnan Province. Nearly full-length HIV-1 genome sequences were obtained from three individuals with no epidemiological link, including two men who have sex with men and one individual with heterosexual contact. These individuals were reported in 2023. These sequences form a monophyletic clade that is distinct from all known subtypes and CRFs. Recombination and phylogenetic analyses revealed that this recombinant strain exhibited a consistent mosaic genomic structure, with the CRF55_01B backbone incorporating two CRF07_BC segments. Bayesian phylogenetic analyses indicated that this recombinant form originated around 2019. In accordance with standard nomenclature, it has been designated CRF200_0755. In addition, this recombinant strain carries the V179E resistance mutation from CRF55_01B. The discovery of this novel CRF highlights the ongoing evolution of HIV-1 amid the co-circulation of prevalent strains and population mobility. It also emphasizes the importance of continuous molecular surveillance in guiding public health strategies, given the potential presence of preexisting resistance mutations.
The persistent emergence of novel circulating recombinant forms (CRFs) of HIV-1 in China is attributed to the prolonged co-circulation of divergent subtypes and CRFs among diverse high-risk populations, facilitating dual infections and recombination events. CRF07_BC first appeared in southwestern China in the early 1990s and initially spread among people who inject drugs. 1 Due to its high transmissibility and reduced virulence, it spread rapidly within sexually active populations. The proportion of CRF07_BC cases increased from 24.8% between 2004 and 2009 to 42.2% between 2020 and 2023, by which time it had become the most prevalent circulating strain in the country.2,3 CRF55_01B, the first CRF identified among men who have sex with men (MSM) in China, originated in Guangdong Province. 4 It experienced rapid growth between 2005 and 2009, followed by sustained expansion after 2010. 5 Due to the high mobility of MSM populations, CRF55_01B spread widely from south to north and from coastal to inland regions. 5 Between 2020 and 2023, it became China’s fifth most prevalent strain, accounting for 3.8% of cases. 2 The concurrent prevalence of these two strains among high-risk populations such as MSM created opportunities for genetic recombination between them.
Yunnan Province, a southwestern border region of China with extensive population mobility and complex transmission dynamics, serves as a hot spot for the emergence of novel HIV-1 recombinants. Molecular epidemiological investigations in Xishuangbanna Prefecture have revealed a high level of genetic diversity of HIV-1 in this border region. Between 2009 and 2011, CRF08_BC replaced CRF01_AE as the predominant genotype, with multiple unique recombinant forms (URFs) being identified. 6 A recent comprehensive survey confirmed the continued dominance of CRF08_BC (41.5%), alongside CRF01_AE (20.1%), CRF07_BC (16.6%) and a higher proportion of URFs (13.1%). CRF55_01B was also detected at a rate of 1.6%. 7 These findings emphasize the region’s ongoing recombination potential. This study reports the identification and genomic characterization of a novel HIV-1 CRF in this region, resulting from recombination between CRF07_BC and CRF55_01B, designated CRF200_0755.
Plasma samples were collected in 2023 from three individuals newly diagnosed with HIV-1 during routine molecular epidemiological surveillance in Xishuangbanna Prefecture, Yunnan Province, a southern border region of China. The surveillance included 313 newly reported cases in 2023, all of which were subjected to subtyping based on three genome segments, gag (HXB2: 781–1861), pol (HXB2: 2147–3462), and env (HXB2: 7002–7541). The three samples selected for near-full-length genome (NFLG) sequencing were those showing discordant regional assignments: CRF55_01B in both gag and pol regions, but preliminarily classified as subtype C in the env region. The participants included two MSMs (sample IDs: 23BN060 and 23BN063) and one individual infected via heterosexual contact (sample ID: 23BN062). Detailed demographic and epidemiological characteristics are provided in Table 1. No epidemiological linkages were identified among them. The study was approved by the Scientific Research Ethics Review Committee of the Yunnan Provincial Centers for Disease Control and Prevention. All participants provided written informed consent. For participants aged below 18, written informed consent was obtained from their legal guardian. Viral RNA was extracted from the plasma samples. NFLG sequences were amplified using nested PCR and Sanger sequencing, as in a previous study. 8 These sequences were submitted to GenBank under the accession numbers PZ147713–PZ147715. The phylogenetic analyses were performed with MEGA12. A phylogenetic tree of NFLG sequences was constructed using the neighbor-joining method based on the Kimura two-parameter model. Phylogenetic trees of subregions were constructed using the maximum likelihood method and a general time reversible (GTR) nucleotide substitution model with gamma distribution (R4) and invariable sites (I). Recombination analysis was performed using SimPlot 3.5.1 and the Recombinant Identification Program (RIP) online tool. Breakpoint positions were refined by phylogenetic analyses of individual subregions. Bayesian phylogenetic analysis was conducted using BEAST V1.10.4 to estimate the time of origin of the strain. Prior to the Bayesian analysis, the temporal signal was assessed using TempEst v1.5.3. A regression analysis was performed on root-to-tip genetic distances against sampling dates. The datasets exhibited a sufficient temporal signal (R2 >0.8) with the mode of best-fitting root, which supports the use of a molecular clock model. Bayesian phylogenetic analysis was conducted using BEAST V1.10.4. The uncorrelated lognormal relaxed molecular clock was employed alongside the Bayesian skyline coalescent tree priors, using the GTR + I + G4 nucleotide substitution model. Each Bayesian Markov chain Monte Carlo analysis was run for 100 million generations, with sampling every 10,000 generations. Convergence was assessed using Tracer v1.7.2, and effective sample sizes >200 for all parameters were considered adequate. The maximum clade credibility tree was generated using TreeAnnotator after discarding the first 10% of states as burn-in.
Demographic Characteristics of HIV-1-Infected Participants

Initial phylogenetic analysis of NFLG sequences revealed that this strain formed a distinct monophyletic branch that clustered adjacent to known CRF_0755 variants, yet remained clearly distinct from all previously identified subtypes and CRFs in China (Fig. 1A). Subsequent RIP (Supplementary Fig. S1) and Bootscan analyses showed that these sequences shared a consistent recombination pattern formed by the recombination of the parental CRF55_01B and CRF07_BC strains. The novel genome used CRF55_01B as a backbone and was divided into five subregions by four recombination breakpoints: ICRF55_01B (709–6456), IICRF07_BC (6457–7364), IIICRF55_01B (7365–7486), IVCRF07_BC (7487–8427), and VCRF55_01B (8428–9506) (Fig. 1B and C). Phylogenetic analysis of each subregion confirmed their parental origins. Subregions I, III, and V clustered with the corresponding regions of CRF55_01B, while subregions II and IV clustered with the corresponding region of CRF07_BC (Fig. 2A). As these sequences lack epidemiological links, they meet the criteria for a novel CRF designation. They are therefore formally named CRF200_0755. Genetically, this recombinant form exhibits gag, pol, vif, vpr, tat, rev, and nef gene regions derived from CRF55_01B and vpu and env gene regions derived from a combination of CRF07_BC and CRF55_01B.
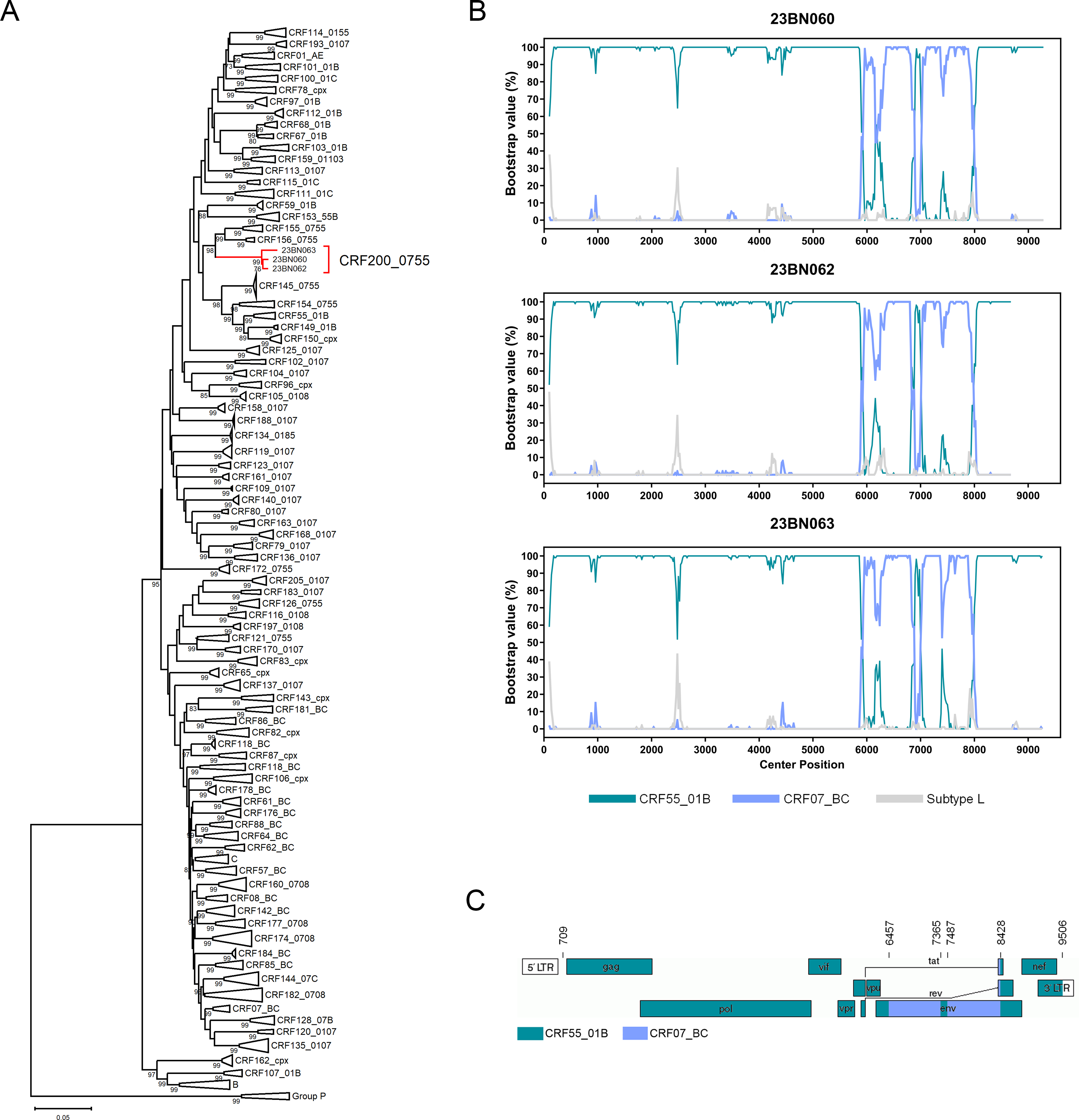
Phylogenetic and recombinant analyses based on the near-full-length genome sequences.
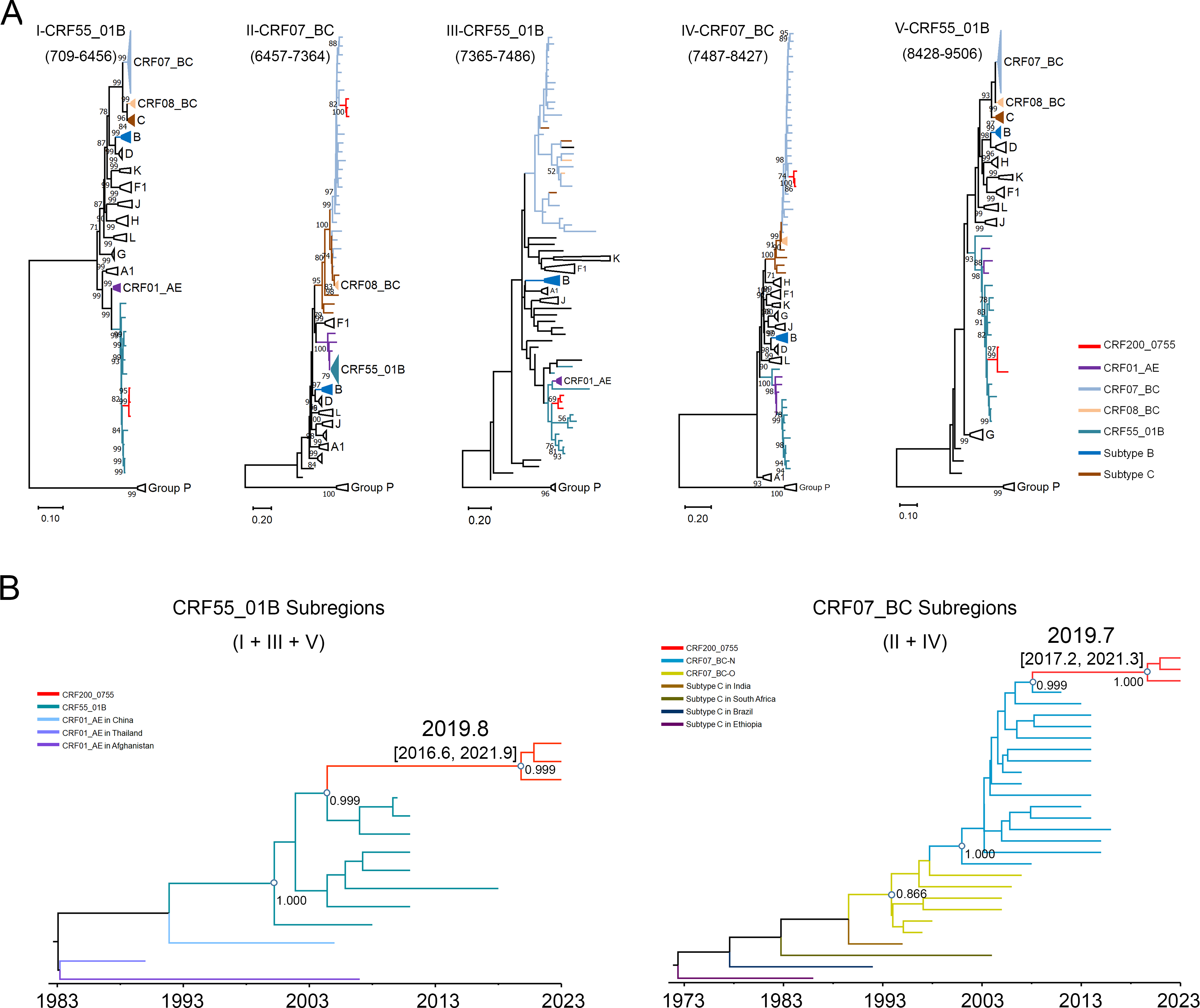
Phylogenetic analysis of individual subregions and Bayesian evolutionary analyses of concatenated CRF55_01B and concatenated CRF07_BC subregions from CRF200_0755.
Further Bayesian phylogenetic analyses were conducted to investigate the origin of CRF200_0755, focusing on the merged CRF55_01B subregions (I + III + V) and the CRF07_BC subregions (II + IV). The results revealed that the median estimates for the time to the most recent common ancestor of the two subregions were 2019.8 and 2019.7, respectively, with 95% highest posterior density intervals of 2016.6–2021.9 and 2017.2–2021.3 (Fig. 2B). These results suggest that CRF200_0755 emerged around 2019.
To date, several novel CRF variants formed by recombination between CRF55_01B and CRF07_BC have been reported in China. These include CRF126_0755, CRF145_0755, CRF154_0755, CRF155_0755, CRF156_0755, CRF172_0755, and CRF200_0755.10–13 With the exception of CRF172_0755, the other CRF_0755 variants primarily utilize the CRF55_01B backbone with inserted CRF07_BC gene fragments. Notably, the breakpoints of CRF200_0755 at positions 6457 and 8426 are close to those of CRF155_0755, CRF156_0755, and CRF172_0755. To investigate whether these shared breakpoints reflect direct phylogenetic relationships, we compared the recombination structures of the four CRFs and performed phylogenetic analyses of the CRF07_BC-derived fragments shared by these four CRFs (6457–7364 and 7487–8397), as well as a CRF55_01B-derived fragment (2426–3390) (Supplementary Fig. S2). The results showed that CRF200_0755 was not directly related to CRF155_0755, CRF156_0755, or CRF172_0755. However, CRF155_0755 and CRF156_0755 were closely related. This suggests that these CRFs originated from independent recombination events, despite their similar breakpoint positions.
These recombination events of CRF07_BC and CRF55_01B did not occur at a single point in time. Their evolutionary origins span from 2005–2007 to 2019–2020.11,12 This indicates that such recombination has been ongoing for nearly two decades. This pattern aligns with previously noted epidemiological trends of CRF07_BC and CRF55_01B. 2 Epidemiological data revealed that these CRFs were predominantly detected among MSM in China, with infected individuals distributed across multiple provinces, including Guangdong, Yunnan, Guizhou, Zhejiang, Sichuan, and Chongqing. Notably, individuals infected with CRF126_0755, CRF156_0755, and CRF200_0755 reported heterosexual contact, suggesting that bridge populations facilitate transmission across different groups.10,12 The samples in this study originated from a tourist city where infected individuals engage in both homosexual and heterosexual contact, indicating the potential for cross-regional and cross-population transmission.
The emergence of these novel CRFs has significant biological and clinical implications. In terms of drug resistance, the V179E non-nucleoside reverse transcriptase inhibitor (NNRTI)-associated resistance mutation originating from the CRF55_01B parental strain was detected in all six CRF_0755 variants except CRF145_0755. 14 According to the Stanford University HIV Drug Resistance Database, V179E is a somewhat polymorphic, NNRTI-selected accessory mutation. In combination with other NNRTI drug resistance mutations, it appears to contribute to a low level of reduced susceptibility to each of the NNRTIs. 15 Furthermore, ongoing genetic recombination substantially increases HIV-1 genetic diversity within MSM populations. This complexity could alter the virus’s biological properties, threatening the accuracy of future diagnostic reagents, vaccine development strategies, and the efficacy of treatment and prevention regimens.
This study reported on a third-generation recombinant form of HIV-1, named CRF200_0755, formed by the recombination of CRF07_BC and CRF55_01B. It was estimated that CRF200_0755 originated around 2019. The emergence of a series of novel CRF_0755 variants, including CRF200_0755, increases HIV-1’s genetic diversity, which poses potential challenges for future prevention and treatment due to preexisting drug resistance mutations and altered viral biology. Therefore, continuous surveillance and investigation of HIV-1 recombinant strains are essential to better understand their evolutionary patterns and biological implications.
Sequence Data
The sequences have been deposited in GenBank (accession numbers: PZ147713–PZ147715).
Authors’ Contributions
M.C., W.D., and M.J. conceived and designed the project. Y.M. and H.C. collected the samples and collected the epidemiological data. M.C. performed the experiments, analyzed the data, and prepared the article. All authors reviewed the article.
Supplemental Material
sj-docx-1-aid-10.1177_08892229261469040 — Supplemental material for Sequence Notes: Identification of a Novel HIV-1 Circulating Recombinant Form (CRF200_0755) in a Southern Border Region of Yunnan, China
Supplemental material, sj-docx-1-aid-10.1177_08892229261469040 for Sequence Notes: Identification of a Novel HIV-1 Circulating Recombinant Form (CRF200_0755) in a Southern Border Region of Yunnan, China by Min Chen, Huichao Chen, Yanling Ma, Wenfei Ding, and Manhong Jia
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.