
Abstract
Background:
Breast cancer is the most frequently diagnosed cancer among Turkish women and both the incidence and associated mortality appear to be increasing. Of particular concern is the percentage of young women diagnosed with breast cancer; roughly 20% of all breast cancer diagnoses in Turkey are in women younger than 40 years. Increased DNA methylation in the promoter region of tumor suppressor genes is a promising molecular biomarker, and human milk provides exfoliated breast epithelial cells appropriate for DNA methylation analyses. Comparisons between DNA methylation patterns in epithelial (epithelial-enriched) and nonepithelial (epithelial-depleted) cell fractions from breast milk have not been reported previously.
Objective:
In the present study, we examined promoter methylation of 3 tumor suppressor genes in epithelial-enriched and epithelial-depleted cell fractions isolated from breast milk of 43 Turkish women.
Methods:
Percentage methylation in the promoter region of Rass association domain family 1 (RASSF1), secreted frizzle related protein 1 (SFRP1), and glutathione-S-transferase class pi 1 was determined by pyrosequencing of the epithelial-enriched and epithelial-depleted cell fractions.
Results:
Pyrosequencing identified a few subjects with significantly increased methylation in 1 or more genes. There was little correlation between the 2 cell fractions within individuals; only 1 woman had increased methylation for 1 gene (SFRP1) in both her enriched and depleted cell fractions. Methylation was positively associated with age for SFRP1 (epithelial-depleted fraction) and with body mass index for RASSF1 (epithelial-enriched cell fraction), respectively.
Conclusion:
Overall, results show that the methylation signals vary between different cell types in breast milk and suggest that breast milk can be used to assess DNA methylation patterns associated with increased breast cancer risk.
Keywords
Well Established
DNA methylation of tumor suppressor genes is considered a promising molecular biomarker for screening increased breast cancer risk and detecting early breast cancers. DNA methylation is considered an early and potentially reversible event in breast cancer etiology.
Newly Expressed
This is the first report investigating DNA methylation profiles in healthy lactating women in Istanbul, Turkey. Of 43 breast milk samples studied, 4 women had significantly increased mean percentage methylation of at least 1 of the 3 tumor suppressor genes (Rass association domain family 1, secreted frizzle related protein 1, and glutathione-S-transferase class pi 1).
Background
Breast cancer is the most frequently diagnosed cancer among Turkish women and in 2006 accounted for roughly one quarter of all female cancers. 1 The incidence of breast cancer in Turkey continues to increase, as does the disease-related mortality. According to the Pan American Health Organization report, Global Burden of Diseases for Turkey, breast cancer moved from the 29th cause of “years of life lost” in 1990 to the 17th cause of years of life lost in 2010. 2 Although the incidence of breast cancer in Turkey is increasing, it remains significantly lower than that of many Western countries, including the United States (age adjusted incidence 33.7/100 000 in Turkey and 90.9/100 000 in the United States). 1 However, the mean age of diagnosis is significantly younger among Turkish women: 52.5 years.3,4 Roughly 20% of all breast cancers diagnosed in Turkey occur in women ≤ 40 years 4 as compared to the United States, where only 6.5% of all breast cancers are diagnosed in women ≤ 40. 5 Early detection is considered key to reducing breast cancer-related mortalities, yet the most successful method of screening, breast mammography, is both less sensitive on dense breasts and expensive, and therefore, it is not recommended for routine screening of young women. 6 A method of accurately assessing breast cancer risk in young women is needed to identify those women who would benefit most from more vigilant screening.
DNA methylation of tumor suppressor genes is considered a promising molecular biomarker for screening increased breast cancer risk and detecting early breast cancers.7-9 Addition of methyl tags to cytosines of cytosine phosphodiester bonded to guanine (CpG) dinucleotides in promoter regions can lead to transcriptional silencing of genes critical for normal cell function. It is important that DNA methylation is considered an early and potentially reversible event in breast cancer etiology. 10 The most direct way of assessing breast cancer risk is by monitoring breast epithelium. However, access to healthy breast tissue is difficult. The most common methods of obtaining breast tissue from healthy nonsymptomatic women—ductal lavage, nipple aspirate, and fine needle biopsy—are invasive and yield small quantities of cells from limited ducts within the breast. The practical difficulty of obtaining breast epithelium from healthy women has spawned an intense interest in research assessing DNA methylation of peripheral white blood cell (WBC) DNA as a proxy for methylation patterns in breast tissue.11-13 Results from analyses of methylation in WBC DNA have been mixed but, overall, indicate that these peripheral cells will be of limited use in assessing methylation in breast epithelium13,14; thus, the search continues for an effective means of assessing breast cancer risk in healthy women.
Exfoliated epithelial cells present in breast milk have been proposed as a relevant source of DNA for monitoring epigenetic alterations associated with increased risk of developing breast cancer.15-17 To date, the few epigenetic studies of cells isolated from breast milk have been conducted with women living in the United States and have focused solely on DNA methylation of the epithelial cell population. Exfoliated epithelial cells represent a small and varying fraction of the total cell population in breast milk; the majority of the cells in milk are leukocytes (white blood cells).16,17 The extent to which the epigenetic signals of the epithelial and nonepithelial cells in breast milk are correlated has not been reported previously. In the present study, we examined DNA promoter methylation of 3 tumor suppressor genes (Rass association domain family 1 [RASSF1], secreted frizzle related protein 1 [SFRP1], and glutathione-S-transferase class pi 1 [GSTP1]) using state-of-the-art pyrosequencing methods in the epithelial-enriched and epithelial-depleted fractions of cells isolated from the breast milk of 43 women residing in Istanbul, Turkey.
Methods
Study Participants and Collection of Milk Sample
This study was approved by the Clinical Ethical Board of Yeditepe University Hospital in Istanbul, Turkey. Forty-three nursing mothers living on the Asian side of Istanbul who visited the Pediatrics Clinic at Yeditepe University for a wellness appointment for their child were asked to provide a milk sample and complete a health and lifestyle questionnaire. The questionnaire included questions regarding history of breastfeeding, parity, age, height, weight, occupation, family history of breast or ovarian cancer, and diet. Each participant was asked to manually pump all the milk she could easily express at 1 time. None of the women used a breast pump to obtain the milk sample they provided. The fresh breast milk was collected in a sterile glass bottle, kept at ambient temperature, and transferred directly to the laboratory within 3 hours of collection. The questionnaire was completed by each participant at the time of the milk donation. The researchers collected the questionnaire forms and returned to the laboratory with the milk samples. The majority of milk samples were expressed in the morning, but no standardization was enforced about time of last breastfeeding session prior to donation. Less than 10% of the milk samples were collected in the evening, and these milk samples were kept cool (4°C) and taken to the laboratory the following morning.
Processing of Breast Milk, Isolation of Epithelial-Enriched and Epithelial-Depleted Cell Populations, DNA Isolation, Polymerase Chain Reaction, and Pyrosequencing
Breast milk was processed and cells were extracted as previously described. 17 Briefly, the fresh (never previously frozen) breast milk was spun at 805 × g for 10 minutes at 24°C. The fat and supernatant were removed and stored in amber glass jars for future chemical analysis. The cell pellet was washed with phosphate-buffered saline (PBS) solution and spun at 450 × g for 5 minutes; this step was repeated twice. The washed cell pellet was then counted and resuspended in degassed PBS and then incubated with EpCam magnetic beads (Miltenyi Biotec, Auburn, California, USA) for 15 minutes at 4°C. Cells were separated into epithelial-enriched and epithelial-depleted cell fractions using a paramagnetic column (Miltenyi Biotec). After cell separation, each fraction was counted using a hemocytometer; the cell fractions were then spun at 450 x g and the cell pellets frozen at −20°C. DNA isolation occurred within 2 weeks of cell isolation and was conducted using QIAmp mini kit (Qiagen, Valencia, California, USA). DNA quantity and quality were assessed using a NanoDrop 8000 Spectrophotometer (Thermo Scientific, Wilmington, Delaware, USA). Bisulfite treatment was conducted following the manufacturer’s protocols for the Epitect bisulfite treatment kit (Qiagen). Polymerase chain reaction amplification and pyrosequencing were conducted as previously described except that only the first 4, 8, and 8 CpGs were included in the pyrosequencing assays for RASSF1, GSTP1, and SFRP1, respectively. 17 Amplified bisulfite-modified DNA from the epithelial-enriched and epithelial-depleted cell fractions of an individual sample were run on the same pyrosequencing plate. Each plate contained a negative (no template) control and a positive cell line control. The mean coefficients of variation based on quadruplicate samples of cell line DNA from 4 separate pyrosequencing plates were 14% for RASSF1, 0.57% for SFRP1, and 4.5% for GSTP1.
Data Analysis
Statistical analysis was performed using STATA 10: Data Analysis and Statistical Software (StataCorp LP, College Station, Texas, USA). For each of the 3 genes—RASSF1, SFRP1, and GSTP1—mean methylation scores were created for both enriched and depleted samples. To obtain consistent mean measures while avoiding censoring of cases due to missing values for higher CpG sequence numbers, means were created using only the first 4, 8, and 8 CpG site values, respectively. The correlation between enriched and depleted methylation values was assessed using simple scatterplots and ordinary least squares regression. The effects of age (< 30 years), smoking and secondhand smoke, overweight (body mass index [BMI] > 25), and obese (BMI > 30) were each assessed through examining the distribution of methylation values across each of these dichotomous covariates, examining the correlation of mean methylation values on each of these variables using bivariate ordinary least squares, and testing for a difference in mean methylation across each variable using pooled t tests. Last, additional analyses, that is, multivariate regression of sample volume against cell counts and enriched or depleted DNA yield, as well as bivariate regressions of sample volume on age, age at first birth, and BMI, were run separately for enriched and depleted samples to confirm the independence of sample volume from these effects.
Results
Participant and Sample Characteristics
Forty-three women residing in Istanbul donated breast milk and completed a health and reproductive history questionnaire. Selected characteristics of the study participants are shown in Table 1. There was a large range in the ages of participants, spanning 24 years, as well as a large range in the ages of the child nursing (Table 1). Roughly 50% of the women were nursing their first child, 30% their second child, and 20% their third or more child (2 women were nursing their fourth child and 1 woman was nursing her fifth child). Of the 38 women who responded to the questions about family history of cancer, 8% reported that a first-degree family member had breast cancer, and 10.5% reported that a first-degree family member had ovarian cancer. Nine percent of women reported that they currently smoked cigarettes, and 32% reported that they were exposed to secondhand smoke.
Study Population Characteristics, Milk Volume, Cell Counts, and DNA Yield among the 43 Participants (N = 43).
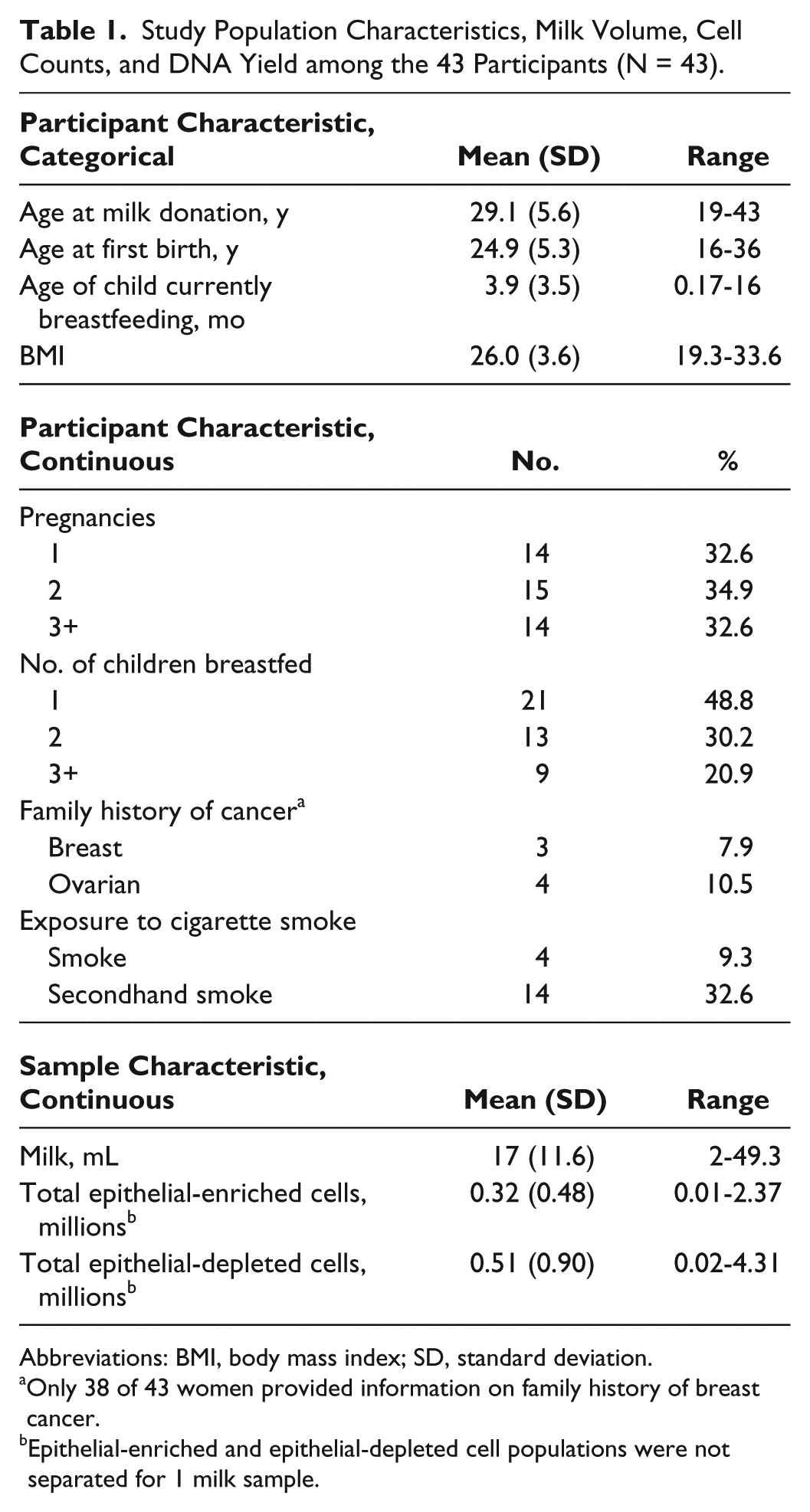
Abbreviations: BMI, body mass index; SD, standard deviation.
Only 38 of 43 women provided information on family history of breast cancer.
Epithelial-enriched and epithelial-depleted cell populations were not separated for 1 milk sample.
The volume of the milk ranged from 2 to 49 mL (Table 1) and was not related to the woman’s age, her baby’s age, or her BMI (overall R2 = 0.089; data not shown). Based on immunomagnetic separation using an antibody to the epithelial cell-specific protein EpCAM, milk samples contained an average of 31 000 epithelial cells/mL (range = 517-328 571 cells/mL) and an average of 303 000 nonepithelial cells/mL (range = 200-4 310 000 cells/mL). Total cell counts are shown in Table 1.
Promoter Methylation of Tumor Suppressor Genes in Epithelial-Enriched and Epithelial-Depleted Cell Fractions
DNA methylation was assessed in the promoter region of 3 tumor suppressor genes: RASSF1 (4 CpG sites), SFRP1 (8 CpG sites), and GSTP1 (8 CpG sites). All pyrograms were inspected, and only those pyrograms where peak heights on the pyrogram were above 50 and the theoretical histogram matched the observed histogram were used to calculate mean methylation scores. Thus, methylation data were available for roughly 40% to 70% of samples for each gene in each of the epithelial-enriched and epithelial-depleted cell fractions (Table 2).
Mean Percentage Methylation Levels across all CpG Sites per Gene.
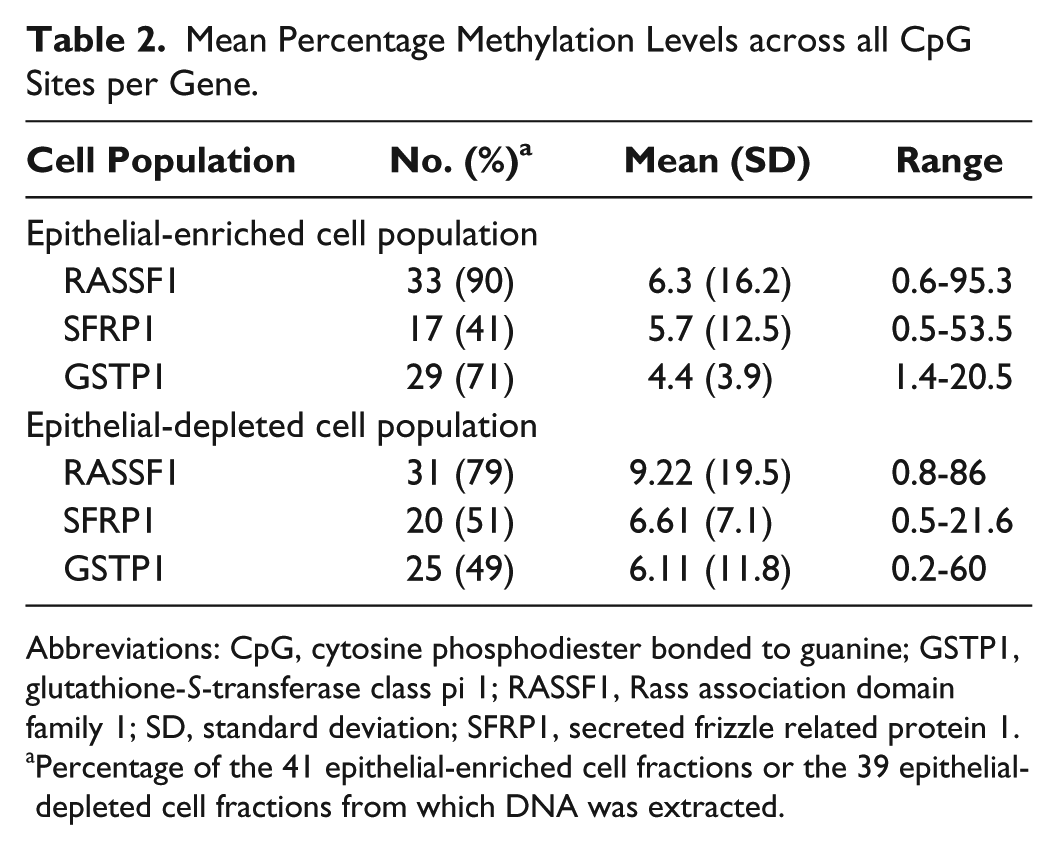
Abbreviations: CpG, cytosine phosphodiester bonded to guanine; GSTP1, glutathione-S-transferase class pi 1; RASSF1, Rass association domain family 1; SD, standard deviation; SFRP1, secreted frizzle related protein 1.
Percentage of the 41 epithelial-enriched cell fractions or the 39 epithelial-depleted cell fractions from which DNA was extracted.
We assessed promoter methylation of tumor suppressor genes in DNA from the epithelial-enriched cell fraction as an indicator of breast cancer risk. As expected of healthy, nonsymptomatic nursing women, the overall mean methylation scores for each of the 3 tumor suppressor genes were low: slightly above 5% for RASSF1 and SFRP1 and below 5% for GSTP1 (epithelial-enriched cell fraction in Table 2 and Figure 1). Nevertheless, 4 DNA samples from the epithelial-enriched cell fraction had mean methylation above 10%, and 1 of these samples was methylated in the promoter region of both RASSF1 and GSTP1—95.3% and 13.4%, respectively. However, none of the donors of these 4 samples had a family history of breast cancer, smoked, or were exposed to secondhand smoke. Three of the 4 donors had BMIs below 30.
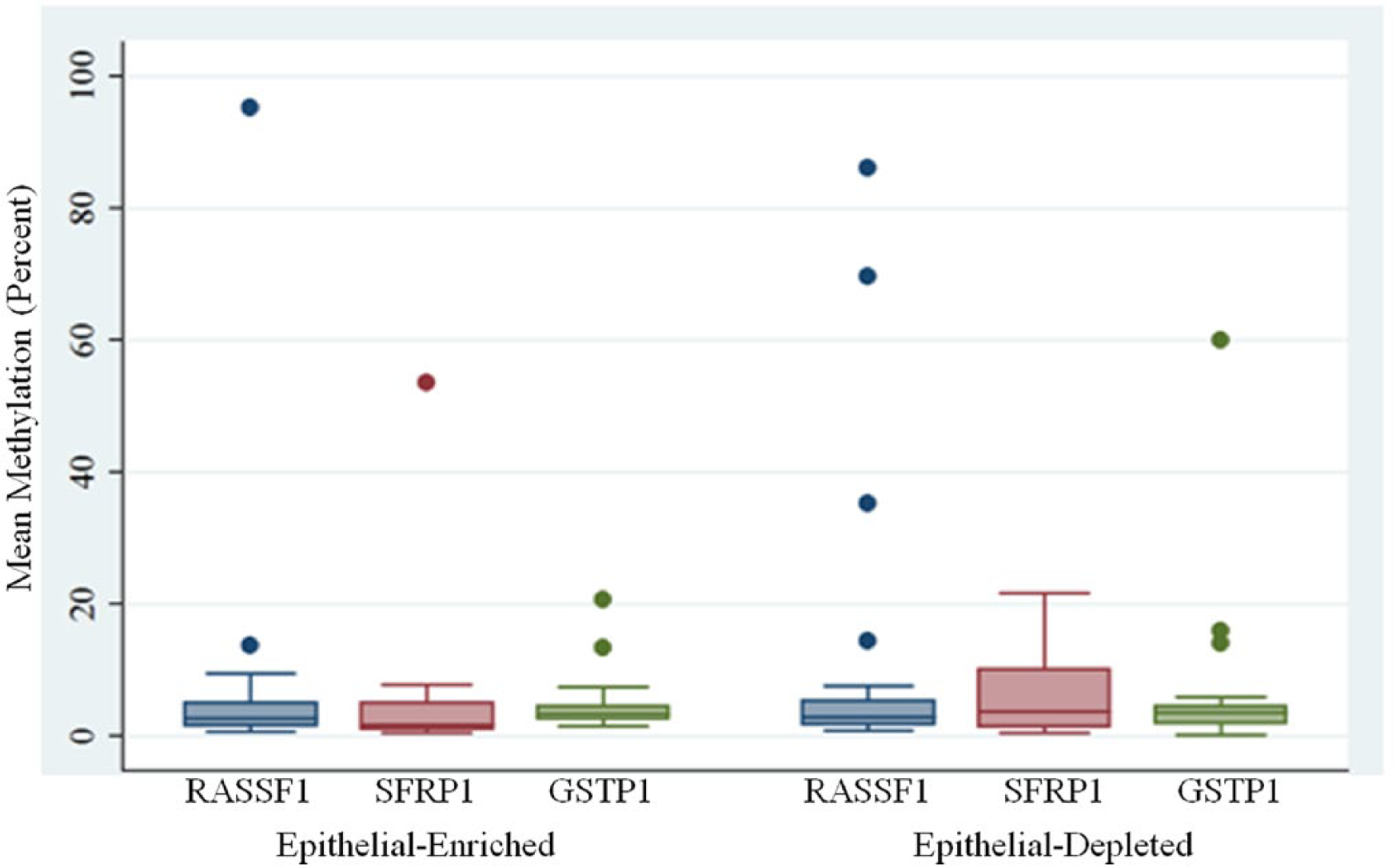
DNA Methylation in Cells from Breast Milk.
As with the epithelial-enriched cell fraction, the mean methylation scores of the epithelial-depleted cell fractions were typically low but slightly higher than the mean methylation scores obtained for the epithelial-cell fraction (Table 2 and Figure 1). Mean methylation scores for all 3 genes were above 5% and several samples had mean methylation values above 10%: RASSF1 (4), SFRP1 (4), and GSTP1 (3).
The degree to which methylation in WBCs can serve as a proxy for methylation in breast epithelial cells is an area of intense research interest.11-13 Because the epithelial-depleted cell population is composed primarily of WBCs, we asked whether mean methylation scores from the epithelial-enriched and epithelial-depleted cell fractions within individuals were correlated. The number of women for which mean methylation scores were available from both cell fractions for each of the 3 genes was limited: 26, 11, and 20 for RASSF1, SFRP1, and GSTP1, respectively. For RASSF1 and GSTP1, there was no correlation between the mean methylation scores obtained from the epithelial-enriched and epithelial-depleted cell populations. In contrast, for SFRP1, the mean methylation scores of the epithelial-enriched and epithelial-depleted cells were significantly correlated (P = .045) and accounted for roughly 38% of the observed variability (Table 3 and Figure 2). However, the significant correlation was heavily dependent on high methylation scores from a single milk sample (epithelial-enriched mean methylation of 53%, and epithelial-depleted mean methylation of 22%), as the SFRP1 correlation disappeared when data from that sample were removed (R2 = 0.03; P = .63).
Correlation between Mean Methylation in Epithelial-Enriched and Epithelial-Depleted Cells in Paired Samples.
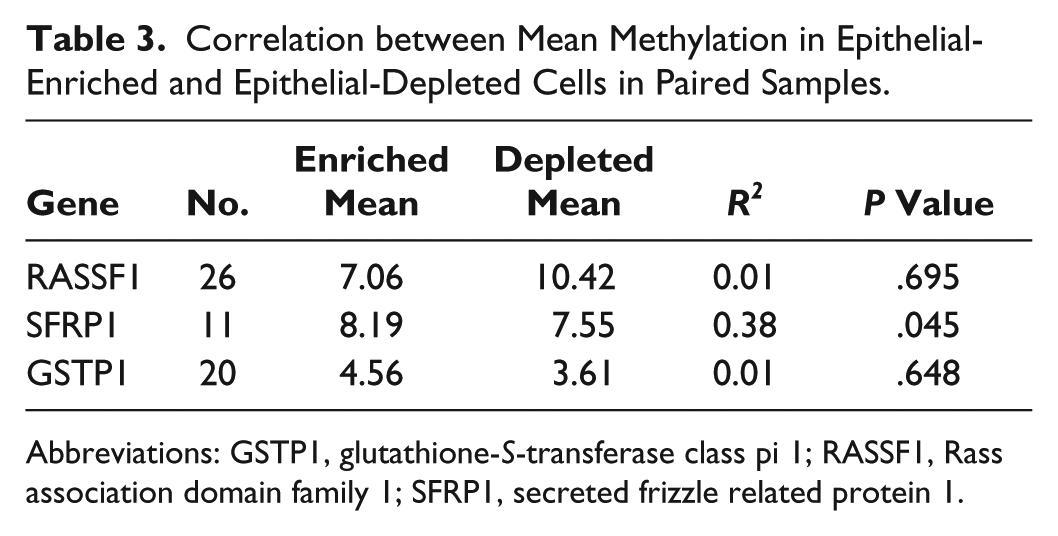
Abbreviations: GSTP1, glutathione-S-transferase class pi 1; RASSF1, Rass association domain family 1; SFRP1, secreted frizzle related protein 1.
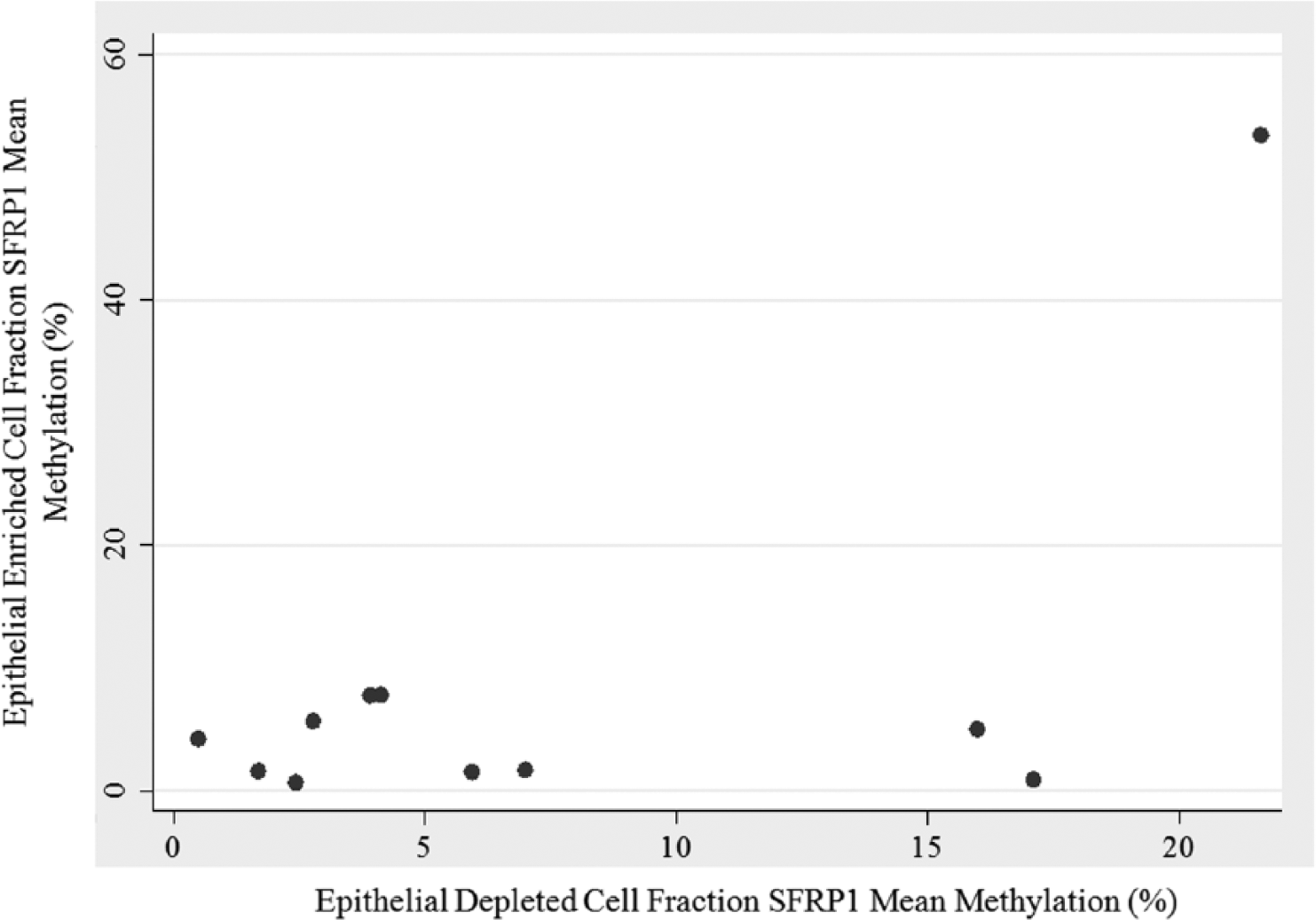
Methylation of SFRP1 in Cellular Fractions of Breast Milk.
Next, we sought to determine if characteristics related to breast cancer risk were associated with increased methylation in the 3 tumor suppressor genes. Due to the small sample size, we could statistically analyze only age and BMI by dichotomizing as follows: young and not young (< 30 and ≥ 30 years of age), and obese and not obese (> 30 and ≤ 30 BMI). Women’s age was found to be positively associated with mean methylation for only 1 of the 3 genes, SFRP1, in the epithelial-depleted cell fraction only (Table 4). The mean methylation score of SFRP1 for women 30 years and older was roughly 4 times greater than the mean methylation score of women younger than 30 years (11.2 vs 2.9, respectively; P = .005). However, the sample size for this comparison was small with only 11 women younger than 30 years and 9 women 30 years or older. Similarly to age, women’s BMI was found to be positively associated with mean methylation for only 1 of the 3 genes, RASSF1, but this time the relationship was detected in the epithelial-enriched fraction only (Table 4). The mean methylation score of RASSF1 for obese women was 6.64 times greater than the mean methylation score of not obese women (26.0 vs 3.9, respectively; P = .016). Again, however, the sample size for this comparison was very small with only 4 women having a BMI greater than 30 and 24 women with a BMI of 30 or less.
Effects of Age and BMI on Promoter Methylation of Tumor Suppressor Genes in DNA Extracted from the Epithelial-Enriched and Epithelial-Depleted Cell Fractions Obtained from Breast Milk.
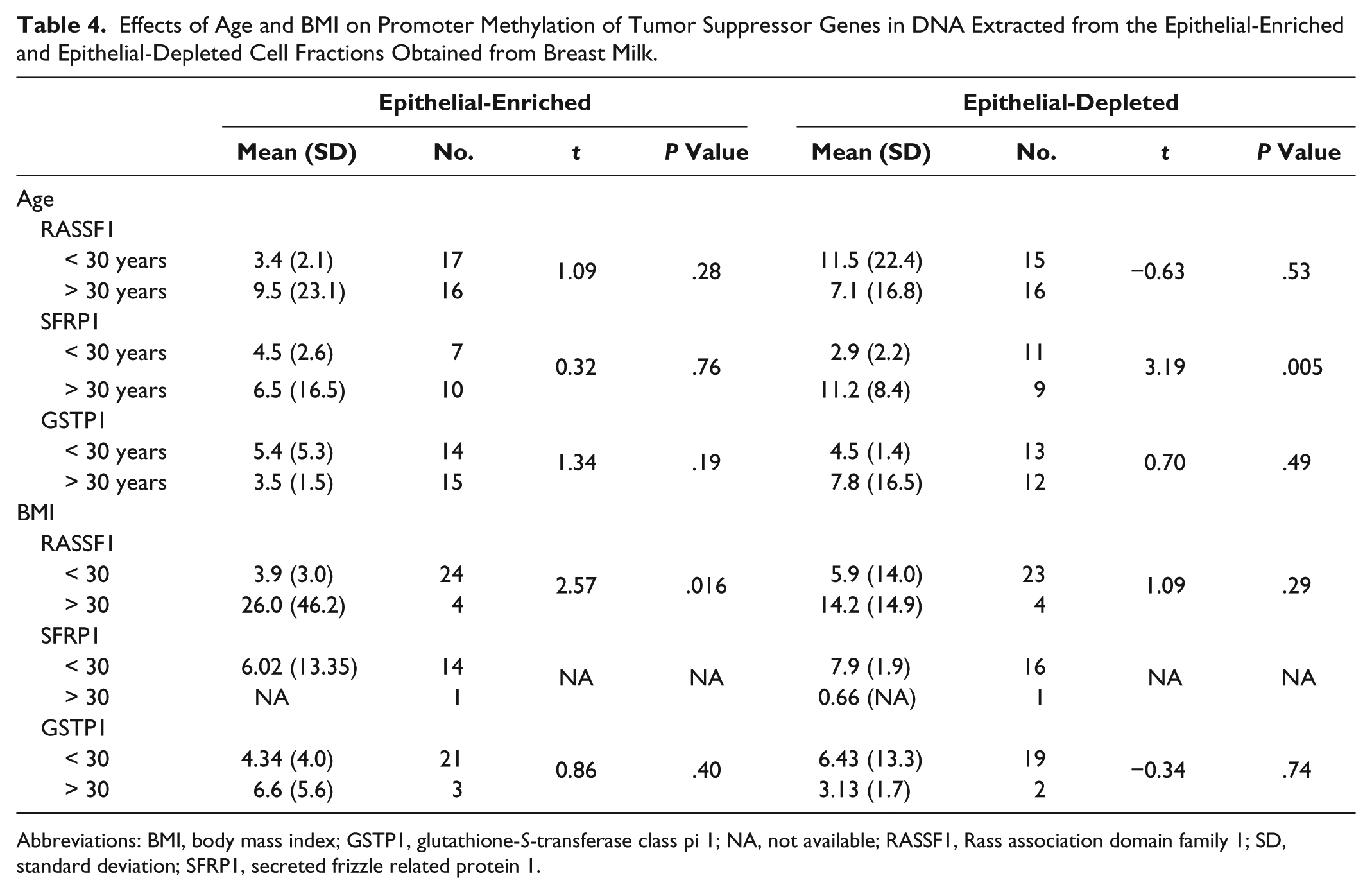
Abbreviations: BMI, body mass index; GSTP1, glutathione-S-transferase class pi 1; NA, not available; RASSF1, Rass association domain family 1; SD, standard deviation; SFRP1, secreted frizzle related protein 1.
Discussion
In our study of 43 lactating women, promoter methylation analysis of 3 tumor suppressor genes in DNA extracted from the epithelial-enriched and epithelial-depleted cell fractions of breast milk yielded mean methylation scores only slightly above 5%, the limit of quantification and lower limit of detection for pyrosequencing. 18 The low methylation values obtained from both cell fractions are expected of healthy nonsymptomatic women. Of significant interest in assessing breast cancer risk are the few high methylation scores in the epithelial-enriched cell fraction. Four women had significantly increased mean percentage methylation of at least 1 of the 3 tumor suppressor genes, and each gene was methylated in at least 1 sample. For each gene, roughly 5% of the samples that passed quality control had methylation values above 10% (RASSF1A 2/33; SFRP1 1/17; and GSTP1 2/29). This level of increased methylation is similar to that observed in the milk of healthy women in the United States (RASSF1A 6/102; SFRP1 11/101; and GSTP1 0/99). 16 The panel of genes—RASSF1, SFRP1, and GSTP1—was selected for their roles in cell cycle, apoptosis, and toxicant metabolism, and because all 3 genes have been shown to be hypermethylated in breast cancer and to be associated with increased breast cancer risk.17,19-24 Thus, our results demonstrate that 4 women (10% of the participants) have a methylation pattern indicative of increased breast cancer risk. The extent to which the present results accurately predict individual risk of developing breast cancer has not been determined. Long-term follow-up, a more extensive gene panel, and a larger sample size are all required to fully evaluate the sensitivity and specificity of methylation signals in breast epithelial cells for predicting breast cancer risk. Still, the results presented here are promising, as access to the target cell population has been a major limiting factor in applying methylation analysis to breast cancer risk.
Breast cancer among Turkish and Asian women peaks between the ages of 40 and 50 years,3,4,25 in contrast to European and North American women, in whom the disease peaks between 60 and 70 years of age.3,26 This discrepancy in age of peak incidence has prompted some to speculate whether breast cancer is the same disease among women of Asian and Western countries 3 and highlights the need for effective methods of screening young women. Further complicating the picture of breast cancer in Turkey are the changes in demographics and risk factors that occur as women adopt a more “Western” lifestyle.27,28 Increased BMI, older age at first full-term pregnancy, and reduced breastfeeding have all been associated with increased breast cancer risk27,29 and are considered part or consequences of the Western lifestyle.
In the present study, we also assessed promoter methylation of tumor suppressor genes in the epithelial-depleted cell fraction and found little evidence that the signals in the 2 fractions were correlated within individuals. This finding indicates (1) that the immunomagnetic bead separation is isolating 2 distinct populations and (2) that the primarily leukocyte population of the epithelial-depleted cell fraction is providing information related to something other than the methylation of breast epithelium. At present, it appears that separation of the cell fractions is required for detecting methylation signals of breast epithelium; however, we did not evaluate unseparated cell populations. Given the cost and time associated with cell separation, it would be ideal if the methylation levels of the total cell population in breast milk were an accurate predictor of malignancy in the breast epithelium. However, the preliminary results of the current study indicate that separation is necessary given that there is no correlation in methylation patterns between epithelial-enriched and epithelial-depleted cell fractions in the 3 tumor suppressor genes analyzed.
One limitation of the present study is the small sample size, especially for subanalyses. The initial sample size of 43 participants was small, but the failure to obtain pyrosequencing results for all samples further reduced the sample size for methylation analyses. Because milk was processed within a few hours of being expressed, we expected all milk samples to yield sufficient quality and quantity of DNA for complete methylation analysis, similar to that reported by Wong and colleagues. 16 However, breast milk volumes collected in the present study from Turkish women were much lower than in the previous study of US women (mean = 17 and 86 mL, respectively). 16 The small volumes yielding low quantities of DNA were the major limitation in pyrosequencing quality. Future studies should include collection of larger volumes of milk and possibly multiple samples to ensure sufficient cell populations and DNA for methylation analysis.
Conclusion
Overall, results show that the methylation signals vary between different cell types in breast milk and suggest that breast milk can be used to assess DNA methylation patterns associated with increased breast cancer risk.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by the Scientific and Technological Research Council of Turkey (TUBITAK Project No. 133S155) and the Avon Foundation.