
Abstract
Objectives:
To review the pharmacology, efficacy, and safety of Brexanolone and define its role in the treatment of postpartum depression.
Date Sources:
A MEDLINE/PubMed search was conducted (1980-May 2020) using the following keywords: postpartum depression, antidepressants, pharmacologic therapy, drug therapy, and brexanolone to identify relevant articles.
Study Selection/Data Extraction:
Literature search was limited to human studies published in the English language. Phase I, II, and III studies evaluating the pharmacology, efficacy, safety of brexanolone for postpartum depression were included. Bibliographies of relevant articles evaluating postpartum depression and treatment were reviewed for additional citations and background information.
Data Synthesis:
Brexanolone is a soluble, proprietary, injectable formulation of allopregnanolone, a neuroactive steroid that modulates neuronal excitability. Allopregnanolone levels increase during pregnancy and decrease substantially after birth. These fluctuations have profound effects on anxiety and depression. Three clinical trials established the efficacy and safety of brexanolone in the treatment of postpartum depression. In all 3 trials, brexanolone had an acceptable safety profile and was well tolerated. The most common adverse effects were loss of consciousness, sedation, dry mouth, headache, dizziness, and flushing. Due to sudden loss of consciousness and excessive sedation, continuous pulse oximetry is recommended.
Conclusion:
Brexanolone has a novel mechanism of action and appears to be safe and effective for the treatment of moderate to severe postpartum depression. At present, high cost, serious adverse effects, and restricted access may limit its use in clinical practice.
Background
Postpartum depression (PPD), also known as postnatal depression, is a serious and debilitating mental disorder. PPD may occur in childbearing females as early as the third trimester, and up to 4 weeks after delivery, as defined in the Diagnostic and Statistical Manual of Mental Health Disorders (DSM-5). 1 PPD affects women from all countries and cultures; based on epidemiologic studies, prevalence is between 8-20% in new mothers. Many women with PPD recover within a few months; about 30% of episodes last beyond the first year of postpartum. Therefore, it can negatively impact the short- and long-term quality of life for new mothers, their children, family, as well as present a significant burden to the healthcare system. Due to the stigma associated with psychiatric disorders, women may hesitate to seek treatment or be diagnosed. In addition, educational disparities, race, ethnicity, and country of origin may restrict access to care.2,3
The precise etiology of PPD has not been fully established, but it appears to be affected by both biologic and environmental factors. Some diagnoses may be related to a previous history of depression or other mental health disorders, especially bipolar disorder or schizophrenia. 3 Other factors such as emotional stressors, social relationships, and financial strain may increase one’s risk. Interestingly, epidemiologic studies have identified that the depletion of maternal long chain omega-3 fatty acids, and significant fluctuations in hormone concentrations (i.e., estrogen and progesterone) may play a role in the pathophysiology of PPD.3-5
Given the prevalence and serious nature of this disorder, assessments should be performed and documented on a regular basis. 6 Although screening can be feasible and is helpful, therapy selection is often complex during pregnancy and postpartum due to liability risks, breastfeeding implications, and maternal morbidity. 7 Therefore, treatment should be individualized and include both nonpharmacologic and pharmacologic approaches. Nonpharmacologic practices, which may alleviate the stress of new parenthood, include good sleep hygiene, exercise, adequate nutrition, support systems (family or professional), cognitive behavioral therapy, and electroconvulsive therapy. As for pharmacologic therapy, antidepressants are considered first-line treatment for PPD. Based on the presumption of depletion in maternal long chain omega-3 fatty acids and hormonal fluctuations, these may also be considered as treatments after evaluating the risks and benefits.8,9
Few studies have compared the different classes of antidepressants used in PPD. Historically, tricyclic antidepressants have been used, with nortriptyline accepted as the safest for pregnancy and breastfeeding.10-15 Currently, selective serotonin reuptake inhibitors (SSRIs) are often first choice due to better adverse effect (AE) profiles and tolerance, while maintaining efficacy. Sertraline has been the most extensively studied and prescribed due to its favorable safety profile in infants exposed during lactation and breastfeeding with over 50% efficacy rates. 12 Other antidepressants with evidence in PPD include venlafaxine, bupropion, and nefazodone.13-15 Although available literature supports the use of antidepressants, their maximal effects are achieved after multiple weeks of treatment. This delay may cause a significant burden to the new mother, family, and the healthcare system. Limitations of available data include a small number of randomized and controlled trials, underpowered samples, lack of control groups, short follow-up periods, and possible risk of bias. 16
Omega-3 fatty acids have gained interest in the general population as well as in the treatment of postpartum depression based on proven health benefits in cardiovascular disease, according to the 2017 American Heart Association guidelines. 17 Studies evaluating omega-3 fatty acids have shown improvement in depression scores. However, these results may be misrepresented due to the supportive psychotherapy arm, lower baseline depression scores, and small sample sizes. Until well designed, adequately powered clinical trials provide optimal dosing and duration of therapy, omega-3 fatty acid supplementation can be considered as adjunctive therapy but should not be recommended as first line therapy for PPD.18-20
Interest in the use of hormonal interventions with PPD is ongoing based on benefits in neuronal growth and survival, as well as reductions in oxidative stress. 9 Studies have evaluated various hormonal formulations, such as oral, transdermal, and parenteral. Synthetic progesterone should not be used in the treatment of PPD due to an increased risk in depressive symptoms. Use of estrogen in women can be considered, but should be avoided in those with an increased risk of thromboembolic events. Long-term use should be cautioned due to an increased risk of endometrial hyperplasia and cancer. Although research on estrogen is promising but preliminary, further studies are required to establish a minimum effective dose and duration of treatment.21-25
Due to the negative impact of PPD on the mother, infant, and family, a pharmacologic agent with a targeted mechanism of action, greater efficacy, and quicker onset of action is desirable. This article will review the clinical trial data of brexanolone, the first approved by the Food and Drug Administration (FDA) for the treatment of PPD, and discuss its place in therapy.
Data Selection
A PubMed/MEDLINE search (1980-May 2020) was conducted using key search terms brexanolone, postpartum depression, antidepressants, pharmacologic therapy. Articles evaluating brexanolone in postpartum depression published in the English language were included. Trials were selected for review of efficacy and safety of brexanolone. The prescribing information was obtained from the manufacturer’s website and ClinicalTrials.gov. Bibliographies of all relevant articles were reviewed for additional citations.
Pharmacology and Pharmacokinetics
Brexanolone (Zulresso®) is a neuroactive steroid γ-aminobutyric acid A (GABAA) receptor positive modulator. The exact mechanism by which brexanolone exerts its antidepressive/mood effects is not fully understood. However, it is chemically identical to endogenous allopregnanolone, a progesterone metabolite which also positively modulates GABAA receptors (Table 1). 26 Following childbirth, plasma concentrations of allopregnanolone rapidly decrease, indicating an association between perinatal hormonal fluctuations and GABAA regulations. Decreased levels of allopregnanolone have been commonly associated with major depression and other psychiatric disorders.
Pharmacokinetic Parameters of Brexanolone. 26

a Metabolites are inactive and do not contribute to the overall efficacy
Dosage and Administration
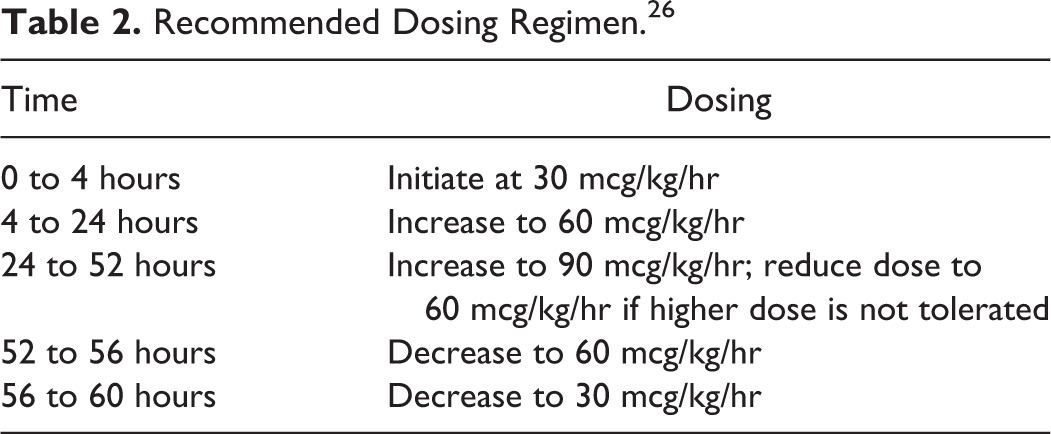
Brexanolone is available as an injectable, sterile, preservative-free, clear, colorless, concentrated solution (100 mg/20 ml) in single dose vials that require dilution (0.9% sodium chloride) prior to administration. The vials should be refrigerated and protected from light. The diluted solution (total volume = 100 ml, 1 mg/ml concentration) is stable up to 12-hours at room temperature and up to 96-hours refrigerated. It is administered over 60-hours (2.5-days) as a continuous intravenous (IV) infusion at titrated rates, depending on patient tolerance, in a supervised clinical setting. The recommended dosing regimen can be found in Table 2. 26
Recommended Dosing Regimen. 26
The effect of brexanolone in infant exposure via breast milk is low due to poor oral bioavailability (studied in 12 women). Safety and efficacy profiles have not been established in pediatrics; brexanolone does not apply to the geriatric population due to the condition discussed. No dose adjustment is necessary in mild, moderate, or severe hepatic and renal impairment; however, brexanolone should be avoided in patients with end stage renal disease.
Cost
The estimated cost of brexanolone is $34,000 per 60-hour course of therapy (estimating an average of 5 vials per treatment). This does not include administration fees or hospital stay.27,28
Adverse Effects
The most common AEs reported in placebo-controlled trials (incidence ≥5%) included loss of consciousness, sedation, somnolence, dry mouth, and flushing. Brexanolone is available through the Zulresso Risk Evaluation and Mitigation Strategy (REMS) Program due to the black box warnings for loss of consciousness and excessive sedation. Patients should be monitored with pulse oximetry every 2 hours during infusion. If excessive sedation is noted, the infusion should be withheld until symptoms resolve; therapy may be resumed at the same or lower dose. Full recovery from loss of consciousness ranged from 15 to 60 minutes after infusion interruption in clinical trials. The pattern and timing of loss or altered consciousness is not clear. If pulse oximetry reveals hypoxemia, immediately stop the infusion and the infusion should not be resumed. 26
Drug Interactions
Studies have not been conducted to evaluate potential interactions involving brexanolone. The key drug interactions reported in clinical trials were associated with use of opioids, antidepressants, or other central nervous system depressants (e.g. benzodiazepines or alcohol). Concomitant use may increase the likelihood and severity of sedation or somnolence. 26
Clinical Studies
Brexanolone received FDA approval based on 4 studies for the treatment of moderate-to-severe PPD in adult females on March 19, 2019. All studies used the Hamilton Depression Rating Scale (HAM-D) scoring to assess depression and response to therapy. 29 These studies include 1 open-label, proof-of-concept study that evaluated safety, efficacy, and pharmacokinetic data in patients with a HAM-D ≥20; 1 phase II trial that evaluated the safety and efficacy profile in patients with severe depression (HAM-D ≥26); and 2 phase III trials that differentiated the efficacy of brexanolone in patients with moderate (HAM-D 20-25) and severe PPD depression (HAM-D ≥26).
Kanes et al 30 conducted an open-label, proof-of-concept study of brexanolone in the treatment of severe PPD. Healthy females 18-45 years of age with a HAM-D score ≥20 and a major depressive episode during the third trimester within 12 weeks postpartum were included; no exclusion criteria was defined. Four out of 6 women screened met the eligibility criteria and completed the study. Baseline mean HAM-D scores were 26.5 ± 4.1 indicating severe depression. Three women had prior history of PPD, 2 also experienced major depressive symptoms outside of the peripartum period. Two were taking sertraline during brexanolone administration, which was allowed per protocol if patients were on a stable dose for more than 14-days prior to screening. The primary outcomes were safety and tolerability measures and secondary outcomes were efficacy assessments (including HAM-D score). Day 1 included a 12-hour dose titration (25%, 50%, and 75% of the maintenance dose for 4 hours at each level). A maintenance dose was continued until 48-hours, where the dose was then titrated down during the remaining 12-hours. Depression symptoms were assessed from the beginning to the end of infusion (60-hours) and a final assessment at 84-hours (initial assessment at hour 12, and repeated at hours 24, 36, 48, 60, and 84). The HAM-D scores decreased and remained low throughout the infusion, mean scores were 1.8 ± 1.5 (p = 0.001 vs. baseline) at the end of infusion and 5.3 ± 2.9 at hour 84. A total of 14 AEs were reported among the patients, including sedation, infusion site reactions, dizziness, flushing, and oral pharyngeal pain. All AEs were mild and self-limited, and none were severe enough to warrant discontinuation. However, 3 patients required AE-related dosage adjustments. According to the authors, brexanolone was well tolerated and demonstrated efficacy in severe PPD. The results of this study are limited by its open-label design, relatively short treatment and assessment period, and a very small sample size.
Kanes et al 31 proceeded to examine the safety and efficacy of brexanolone in the first phase II randomized, double blind, placebo-controlled trial in severe PPD (HAM-D score ≥26). Twenty-one females (aged 18 to 45 years) from 4 U.S. hospitals were included (≤ 6 months postpartum) to receive either a single, IV infusion of brexanolone (n = 10) scheduled at 30 mcg/kg/hr (0-4 hr), 60 mcg/kg/hr (4-24 hr), 90 mcg/kg/hr (24-52 hr), 60 mcg/kg/hr (52-56), and 30 mcg/kg/hr (56-60) or placebo (n = 11). All subjects were in good physical health and had negative pregnancy tests at screening and day 1 of study drug infusion. Patients were excluded if they had active psychosis, suicidal attempts, history of seizures, bipolar or schizoaffective disorders, and history of alcohol or drug abuse in the 12 months prior to screening. Baseline characteristics were similar in both groups, with the exception of a higher history of anxiety in the placebo group (2 vs. 5). Stable antidepressants were acceptable, but initiating new psychotropic therapy was restricted until both the drug infusion and the 72-hour assessment were completed. The primary end-point was the change in HAM-D score from baseline to the end of treatment (60-hour infusion). Secondary endpoints included changes in HAM-D score during follow-up from baseline at 2-hours up to 30-days. Frequent monitoring and assessments were performed to evaluate rapid onset of symptom improvements at 2, 4, 8, 12, 24, 36, 48, 60, and 72 hours, as well as days 7 and 30. At the end of the 60-hour infusion, mean reduction in HAM-D from baseline was 21-points in the brexanolone group compared to 8.8 points in the placebo group (95% CI, -20.77 to -3.67; p = 0.0075). Frequently reported AEs were reported in both brexanolone and placebo groups, such as dizziness (n = 2 vs. n = 3, respectively) and somnolence (n = 2 vs. n = 0, respectively); none of the patients discontinued therapy. Despite the authors interpretation of the findings that brexanolone resulted in a significant and meaningful reduction in HAM-D score, this study had its limitations. These limitations include: strict definition of severe PPD (HAM-D ≥ 26 points), respondent fatigue during follow-up due to frequently administered assessments, and inability to stratify patients due to small sample size.
Meltzer-Brody et al 32 conducted 2 phase III, multicenter, double-blind, randomized, placebo-controlled trials to evaluate the safety and effectiveness of brexanolone for moderate and severe PPD. Women 18-45 years of age meeting DSM-IV criteria for PPD with onset of symptoms during the third trimester or within 4-weeks of delivery were enrolled. Study 1 included 138 patients with HAM-D score of ≥26 and were randomized 1:1:1 to one of the 3 treatment groups: brexanolone 90 mcg/kg/hr (BRX90), brexanolone 60 mcg/kg/hr (BRX60), or placebo. Study 2 randomized 108 patients with HAM-D score of 20-25 to BRX90 or placebo in a 1:1 ratio. Demographic and baseline characteristics were similar in both studies, which included 65% White, 34% Black, and 18% Hispanic or Latino. The average age in the brexanolone group was 28 years and 76% of women reported PPD symptoms within 4-weeks after delivery. Baseline antidepressant use was reported in 23% of patients; however, initiation of new psychotropic medications were restricted. Other exclusion criteria listed were renal failure, hepatic failure, anemia, allergies to allopregnanolone, active psychosis, exposure to another investigative medication, as well as exclusion criteria listed in the previous trial. 29 A titration schedule similar to previously discussed studies was used. The primary efficacy endpoint was a significant mean change in HAM-D score from baseline to end of infusion (hour 60). The secondary efficacy outcome was a mean change in HAM-D at day 30. Both trials showed the following reductions in HAM-D scores: study 1, BRX60 19.5 points (p = 0.0013), BRX90 17.7 points (p = 0.0252), and placebo 14.0 points; study 2, BRX90, 14.6 points vs. placebo 12.1 points (p = 0.0160). In addition, HAM-D score reduction with brexanolone groups at 48-hours was maintained through follow-up on day 30 in study 1 and day 7 in study 2. HAM-D scores were maintained at 19.5 points at both 60 hours and 30 days for study 1, while study 2 HAM-D scores were maintained at 17.7 points for 60 hours and 17.6 points for 30 days. Brexanolone was well tolerated in both studies. Treatment groups experienced similar AEs, which included dizziness, headache, and somnolence. Of note, dose reductions or AE-related interruptions were higher in brexanolone vs. placebo (7% vs. 3%, respectively). In study 1, 2 patients in BRX60 reported serious AEs; 1 patient had suicidal ideation and an intentional overdose attempt, while the other reported somnolence and loss of consciousness. In study 2, 1 patient in BRX90 also reported serious AEs including altered consciousness and syncope. All AEs were considered treatment related.
The authors of this study noted a few limitations in their conclusion. Narrowing participants to moderate and severe symptoms restricted generalizability to a wider population, while there may have also been respondent fatigue due to frequent administrations of HAM-D assessments. The lack of long-term efficacy data is also concerning due to a short follow-up period of 30 days. Based on the author’s interpretation, findings from this trial confirm results from previous phase II data, providing strong evidence in the efficacy and safety of brexanolone in women for the treatment of moderate to severe PPD.
Clinical Considerations
Brexanolone showed significant and clinically meaningful HAM-D score reductions in clinical trials and can be used in patients with moderate to severe PPD. This medication has a rapid onset of action and is given as an IV infusion over a total of 60 hours as a 1-time treatment, which may be appealing to some patients.
Although brexanolone is effective, well-tolerated, and has a quicker onset of action when compared to oral antidepressants, it may not be as safe and is more expensive (at least $34,000) than antidepressants. Barriers to brexanolone use include severe adverse effects requiring pulse oximetry monitoring, administrative costs, lack of long-term safety and efficacy data, absence of head-to-head studies against antidepressants, and time spent away from the newborn. Furthermore, brexanolone access is restricted due to its categorization as a Schedule IV medication, requirement to be enrolled in the Zulresso REMS program, and the need for hospitalization. The clinical studies only included women with moderate to severe PPD, and excluded those with a history of schizophrenia, bipolar disorder, and schizoaffective disorder. Use in patients with mild PPD, suicidal ideation, or a history of psychosis is not recommended.
Questions still remain in regards to the utilization of this agent. The total medical cost, duration of therapy and infusion, and REMS requirement may limit prescribing patterns. With a follow-up period in trials of only 30 days, it is difficult to determine if patients in the brexanolone group experienced any remission or relapse of PPD. It is also difficult to project other long-term safety concerns.
Conclusion
Several clinical trials involving brexanolone are currently ongoing. One phase III study is currently evaluating the safety and efficacy of brexanolone in adolescent females (age 15-17). 33 Another phase III clinical trial investigating the efficacy of allopregnanolone, an oral formulation with high oral bioavailability and once-daily dosing, in major depressive disorder. 34 Results from these trials may expand treatment options and provide insight for future indications. The approval of brexanolone now provides a novel treatment for new mothers who suffer from PPD.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.