
Abstract
Sickle cell disease (SCD) is a hematological disorder that primarily affects individuals of African descent from sub-Saharan Africa and along the mediterranean. The main complications leading to hospitalizations include vaso-occlusive crises (VOCs) and acute chest syndrome (ACS). Therefore, the main objective of this paper was to identify and evaluate evidence-based management and prevention of VOCs in patients with SCD. A literature search of PubMed, Medline Cochrane and Google Scholar database (January 1985 to April 2020) was performed using the following search terms “vaso-occlusive crises”, “sickle cell disease”, “hydroxyurea”, “L-glutamine”, “voxelotor”, “crizanlizumab”, “treatment” and “prevention” as well as a combination of these terms. All English-language interventional studies assessing the efficacy and safety of VOC outcomes were evaluated. Literature was excluded if published in a language other than English or if it was a review article. A total of 69 articles were identified and there were 7 articles that met the search criteria. Majority of the studies focused on mean and median annual rates of VOCs as primary outcomes while median time to first sickle cell crises, median rates of hospitalizations etc were evaluated as secondary outcomes. After reviewing the literature, many patients with VOCs will still benefit from hydroxyurea therapy since long term efficacy data and cost is still a concern for the newer agents including L-glutamine, voxelotor and crizanlizumab. Other factors such as cost or compliance may also be taken into consideration when making recommendations for therapy.
Introduction
Sickle cell disease (SCD) is a hematological disorder that is prevalent in individuals of African descent from sub-Saharan Africa, Saudi Arabia, India, Turkey, Italy, Greece and Spanish speaking areas of Central America, South America and the Caribbean. 1 The Centers for Disease Control and Prevention (CDC) currently does not have an accurate number of patients living in the United States with SCD but it is estimated in 2020 to be around 100,000. 1 They also estimated that 1 out of every 365 African American children will be born with SCD and 1 out of every 13 African American children will be born with sickle cell trait in the United States. 1 In the United States, SCD has a high economic burden costing the healthcare system about $1 billion annually, while indirectly contributing to lost productivity, lower quality of life, premature mortality and uncompensated care.2-4
SCD is caused by a substitution of an amino acid on the beta-globin chain of adult hemoglobin (HbA) from glutamic acid to valine at the sixth position.5,6 This substitution produces sickle-shaped red blood cells through polymerization of oxygen-poor sickle hemoglobin (HbS) leading to multiple complications including vaso-occlusive crises (VOCs), pulmonary hypertension, chronic pain, ischemic complications, anemia, pneumococcal infections and acute chest syndrome (ACS). 5 The 4 main types of genotypes in the United States are HbSS, HbSC, sickle cell β-thalassemia (HbSβ+-thal and HbSβ0-thal).7,8 Patients with the HbSS and HbSβ0-thal genotypes generally present with more severe hematologic and clinical symptoms, while patients with genotype HbSC generally present with milder symptoms but have a higher propensity for developing proliferative sickle retinopathy (PSR).7-9
The common clinical signs and symptoms associated with HbSS include chronic anemia and pallor; fever, arthralgia, scleral icterus, abdominal pain, weakness, anorexia, fatigue, enlargement of the liver, spleen, and heart; and hematuria. Laboratory findings include low hemoglobin levels around 6 to 9 g/dL (60-90 g/L; 3.72-5.59 mmol/L), elevated reticulocytes of 10% to 25%, elevated platelet, elevated white blood cell counts, and normal mean corpuscular volume. The peripheral blood smear demonstrates sickled red cell forms.7,8,10 Currently, the main complications that lead to frequent hospitalizations for SCD are VOCs and ACS.
VOC is an acute complication that affects almost all patients with SCD at some point during their lifetime. 11 VOC usually manifests as acute severe pain affecting 2 or more sites, mainly occurring in the extremities, chest, abdomen, and back.12-14 It is essential to determine the etiology of the pain by obtaining a complete history, physical and pertinent labs, and to rule out other potential causes of pain. 15 Currently, there are no lab tests to rule in or rule out a VOC. 15 Additionally, no biomarkers or imaging studies can validate the pain severity experienced due to a VOC. 15 The lack of objective tests to diagnose VOC makes it essential to utilize the patients self-reported pain scores to guide therapy management.
Patients with an uncomplicated VOC that does not require a hospitalization should not undergo routine laboratory testing; however, they should be inspected for clinical evidence of precipitating factors such as dehydration and infection. 15 Routine labs ordered in patients reporting symptoms of VOC warranting a hospitalization include complete blood count with differential, a reticulocyte count, and a complete metabolic panel including liver function tests. 15 Patients presenting with fever should undergo workup for a possible source of infection including a chest radiograph, urine, sputum, and blood cultures.12,15 The patient’s blood should also be typed and screened proactively in case a need for transfusion arises. 12
For the purposes of review we shall focus on the medical management and prevention of VOCs in patients with SCD, but the focus does not include transfusions or management of pain syndromes associated with VOCs as that has been delineated clearly in multiple publications.12,14-16 Evidence-based guidelines for the treatment of SCD is supported by a panel of experts, and includes a strong recommendation for the use of hydroxyurea in management of these patients. 5 There are limited treatments available for individuals experiencing VOCs with SCD despite the advancements in disease knowledge over the past few decades. Currently, there are 4 FDA approved drugs to manage and prevent complications associated with sickle cell disease. The mainstay of therapy for the past 2 decades was hydroxyurea since its approval in 1998 for SCD, the next agent, L-glutamine (Endari) received FDA approval in July 2017 and the 2 last agents crizanlizumab (Adakveo) and voxelotor (Oxbryta) were both FDA approved in November 2019. Therefore, the objective of this paper is to review the safety, efficacy and place in therapy of these 4 agents in the management and prevention of VOCs in patients with SCD.
Literature Search
A literature search of PubMed and Medline was performed using the following search terms “vaso-occlusive crises”, “sickle cell disease”, “hydroxyurea”, “l-glutamine”, “voxelotor”, “crizanlizumab”, “treatment” and “prevention” as well as a combination of these terms. English language randomized controlled trials in humans assessing safety and efficacy of voxelotor, crizanlizumab, hydroxyurea, l-glutamine published between 1985 to April 2020 were evaluated. A total of 69 articles were identified, of which 7 met inclusion criteria and were utilized for this review. Additional online searches via Google Scholar, Lexicomp, Centers for Disease Control and Prevention (CDC), and package inserts were conducted for updated guidelines, prescribing information and cost information. Bibliographies of selected articles were reviewed manually for relevant publications that focused on the management and prevention of VOCs in adults with sickle cell disease. Review articles (i.e. non primary literature sources) or articles in languages other than English were excluded. A summary of the results is outlined in Tables 1 and 2.
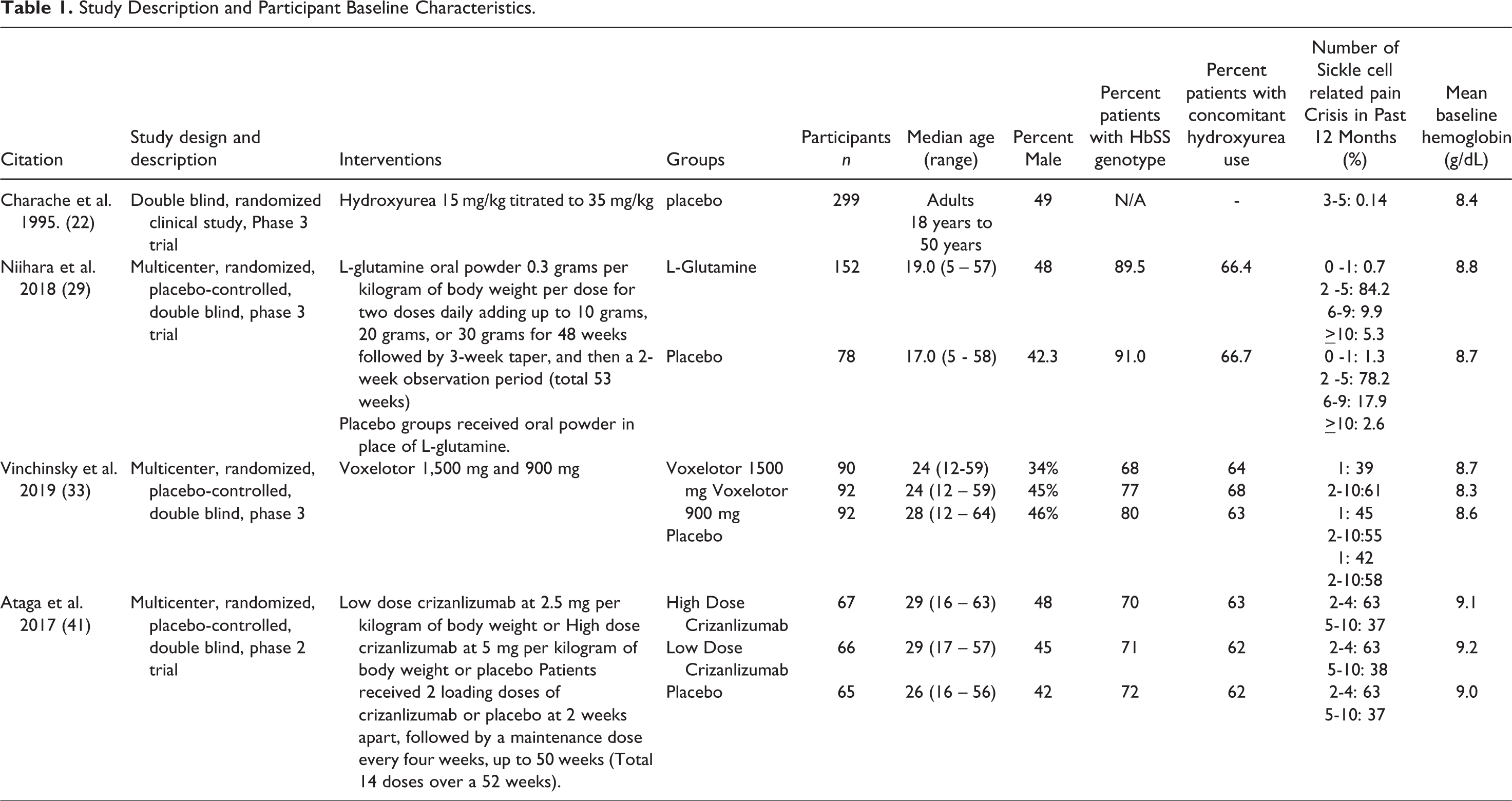
Study Description and Participant Baseline Characteristics.
Summary of Efficacy Outcomes.

* Rate ratio of pain crisis per 48 weeks calculated by dividing the rate amongst patients in the L-glutamine group by the rate among patients in the placebo group.
** Sickle cell related pain crisis defined as an acute episode of pain with no medically determined cause other than a vaso-occlusive event, resulting in a visit to a medical facility and was subsequently treated with oral or parenteral narcotic agents or parenteral nonsteroidal inflammatory drugs.
† Hazard ratio.
Hydroxyurea
Hydroxyurea has been the mainstay of medical management and prevention of VOCs for the past 2 decades after receiving FDA approval in 1998. 17 It is currently indicated for patients who have 3 or more VOCs in the preceding 12 months due to its cytotoxic nature. 18 The mechanism of action in SCD is not fully determined, however it is hypothesized to cause a change in genetic expression of the beta globin. 18 The malformation promotes an increased production of fetal hemoglobin (HbF), typically at least 2-fold above baseline. 18 This effect causes an overall reduction of HbS since hydroxyurea does not affect the formation of the gamma globin chain, the key determinant to produce HbS. 18
HbF is a form of hemoglobin that is normally produced during fetal development and early life. 19 Compared to normal hemoglobin, HbF possesses slightly higher oxygen affinity, however it is clinically well-tolerated and does not pose any risk for problems. 19 With the reduction of HbS and the increase HbF, hydroxyurea leads to less hemoglobin polymerization, altering the adhesion of red blood cells (RBCs) to endothelium, ultimately reducing sickling and improving outcomes. 19 Hydroxyurea is also believed to reduce sickling by increasing nitric oxide production, improving red blood cell rheology, and decreasing neutrophil adhesivity to the vascular endothelium.20,21
The landmark open-label, randomized clinical study of hydroxyurea assigned patients to 2 groups. 22 The first group was assigned to hydroxyurea and the second group was assigned to a placebo. The hydroxyurea group received an initial dose of 15 mg/kg which was titrated up by 5 mg/kg every 12 weeks. If a patient underwent bone marrow depression, treatment was stopped and dose was resumed at 2.5 mg/kg lower than the dose that caused the marrow depression and then continued for another 12-week cycle. All patients concomitantly received 1 mg of folic acid daily and were evaluated every 2 weeks thereafter. 22
The results of this study demonstrated that patients assigned to receive hydroxyurea experienced lower annual rates of crises than those receiving placebo (2.5 vs 4.5 crises per year, P < 0.001). 22 The median time to first crisis was also significantly shorter among patients receiving hydroxyurea (3.0 vs 1.5 months, P = 0.01) and did not cause any significant adverse effects. 22
Fourteen patients had their treatment permanently stopped while almost all in the hydroxyurea group had it temporarily stopped due to myelosuppression with recovery seen within 2 weeks and then later resumed at a lower dose. 22 Approximately, 12 patients had reportedly high hemoglobin levels exceeding 12.8 g/dL and about a similar number in both groups reported high platelet counts of > 800,000 mm3. 22 However, no morbidity associated with high hemoglobin and platelets were found. The most common adverse events included hair loss, rash, fever and gastrointestinal disturbances with similar rates in both groups. 22 Ten patients or their partners became pregnant during the course of study requiring cessation of treatment, and resulting in no adverse consequences to the live born baby at follow-up. 22 Additionally, the main limitation to using hydroxyurea is its potential for teratogenicity and propensity to cause myelosuppression. 17
In 2010, Voskaridou and colleagues published the longest efficacy and safety of hydroxyurea in patients with sickle cell disease. 23 Thirty-four patients with sickle cell anemia HbSS, 131 with HbSβ0-thal, and 165 with HbSβ+-thal participated in this 10-year prospective observational study. 23 The group reported the efficacy of hydroxyurea on the aforementioned genotypes studied was retained after 10 years. The patients who received hydroxyurea had a survival rate of 86%, compared to 65% for patients who did not (P = .001). 23 The levels of fetal hemoglobin at baseline and percentage change of lactate dehydrogenase levels from baseline and at 6 months were the predictors for survival among patients receiving hydroxyurea. This study informed and emphasized the long-term benefit of using hydroxyurea in patients with HBSS, HbSβ0-thal and HbSβ+-thal. 23
L-Glutamine
In July 2017, the FDA approved Endari, a powdered formulation of L-glutamine for use in patients 5 years or older with SCD. 24 L-glutamine is an amino acid that is indicated to reduce the acute complications of SCD. 24 Sickle RBCs are known to be more susceptible to oxidative damage than normal red blood cells. 24 Nicotinamide adenine dinucleotide (NAD+) and its reduced form NADH, function as oxidation-reduction cofactors in red blood cells, which are important in regulating and preventing oxidative damage in RBCs.25,26 L-glutamine is required to synthesize NAD, with greater uptake in sickle red blood cells. 27 The ratio of NADH/(NAD+ + NADH) is lower in sickle red blood cells compared to normal red blood cells, facilitating the chronic hemolysis and vaso-occlusive events experienced by patients with SCD.25,26 L-glutamine exerts its therapeutic effects by targeting the oxidative stress phenomenon present in patients with SCD by increasing the NADH/(NAD+ + NADH) redox ratio.24,28
The pharmaceutical grade L-glutamine formulation obtained FDA approval based on the results of a 53 week phase III safety and efficacy study in patients 5 years or older with HbSS or HbSβ0-thal. 29 Additionally, patients had to experience at least 2 pain crisis in the past year requiring parenteral narcotic or ketorolac in the emergency department, outpatient treatment center, or during hospitalization. 29 Patients randomized to the treatment group received 2 weight based doses daily of approximately 0.3 grams per kilogram of L-glutamine powder per dose adding up to 10 grams, 20 grams, or 30 grams of L-glutamine oral powder daily. 29 This was followed by a 3 week taper, and then a 2 week observation period. Patients in the control group received a placebo powder in place of L-glutamine. Each dose of the powder was mixed with a drink and consumed immediately. 29
Fewer pain crises occurred in patients in the L-glutamine group than placebo groups through week 48 (3.2 vs 3.9, P = 0.005). Patients in the L-glutamine group also had a lower number of hospitalizations for sickle cell related pain, P = 0.005; spent less cumulative days in the hospital, P = 0.02; and had longer time to first and second pain crises when compared to the placebo group. 29 The pain crisis rate ratio in this trial was similar among patients continuing hydroxyurea and those that were not concomitantly receiving hydroxyurea. 29
The rates of adverse events were higher in the L-glutamine group than placebo groups. 29 The majority of adverse events in both groups were gastrointestinal. The most commonly reported adverse events in the L-glutamine group were constipation (25.2%), nausea (22.5%), headache (21.2%), pain in extremity (15.9%), vomiting (14.6%) and non-cardiac chest pain (13.9%), back pain (13.2%) and upper abdominal pain (10.6%). Other adverse events reported in the L-glutamine group at an incidence of less than 10% included diarrhea, nasal congestion, urinary tract infection, fatigue, dizziness and tachycardia. 29 The main limitation to using L-glutamine is the gastrointestinal adverse events. 24
Voxelotor
In November 2019, the FDA approved voxelotor (Oxbryta) for the treatment of SCD in adults and pediatric patients 12 years of age and older. 30 Because deoxygenated Hb is the main driver for HbS polymerization and causing sickle cell crisis, increasing the proportion of oxygenated HbS may be able to circumvent the occlusion. 31 Voxelotor increases oxygen affinity and stabilizes HbS in their oxygenated forms, resulting in less HbS polymerization and occlusion. The therapeutic trials of voxelotor have been studied and found to have favorable results. 31 A phase I, open-label dose-defining study showed all patients receiving multiple doses of voxelotor 500 mg, 700 mg, 900 mg, and 1,000 mg/day for more than 28 days attained clinical improvements in hematological outcomes including increased HbS concentration, reduction of sickle red cells, and hemolysis. 32 Tissue hypoxia and serious adverse events were not observed. 32
The positive impact of voxelotor in preliminary studies led to the pivotal phase III trial to examine the efficacy and toxicity of voxelotor compared to placebo in sickle cell disease patients (HOPE trial). 33 The phase III HOPE trial is a multicenter, international, randomized, double-blind, placebo controlled trial that assigned patients to receive once-daily oral dose of 1,500 mg, 900 mg of volexetor or placebo. 33 The outcomes included the percentage of patients who sustained a hemoglobin response, change in hemoglobin level more than 1g/dL from baseline to week 24. 33 The results demonstrated that 1,500 mg of voxelotor significantly increased hemoglobin concentration in a dose dependent fashion (increase of over 1 g/dL in 51%, 33%, and 7% in the voxelotor 1500 mg, voxelotor 900 mg, and placebo arms, respectively). Markers of hemolysis were also decreased with voxelotor treatment. 33 A post hoc analysis found that episodes of VOCs also significantly reduced, and did not increase with higher doses (annual incidence, 2.8 in both voxelotor doses vs 3.2 in the placebo arms. respectively). 34
Twenty-six percent, 15% and 22% of patients reported headache with the high dose, low dose and placebo respectively. Twenty percent, 17% and 10% of patients reported diarrhea and nausea with the high dose, low dose and placebo respectively. Other side effects that were reported at an incidence of 10% or higher for all 3 groups included arthralgia, upper respiratory tract infections, abdominal pain, fatigue, rash, pyrexia, pain in extremity, back pain and vomiting. There were also similar rates of incidence of adverse events of all grades reported in all 3 groups at 94%, 93% and 89% respectively. 33 The main limitation to using voxelotor is the gastrointestinal adverse events. 30
Crizanlizumab
In November 2019, the FDA approved crizanlizumab-tmca (Adakveo) to reduce the frequency of VOCs in adults and pediatric patients aged 16 years and older with SCD. 35 Crizanlizumab-tmca is a humanized monoclonal antibody that exerts its effects by binding to P-selectin and blocking the interaction with P-selectin glycoprotein ligand 1 (PSGL-1). 35 P-selectin is found in resting endothelial cells and platelets. Upon activation of the cells during processes such as inflammation, P-selectin is transferred to the cell membrane surface where it mediates abnormal rolling and static adhesion of sickle erythrocytes to the vessel surface and vascular occlusion.36-38 P-selectin also triggers activated platelets to bind to neutrophils. 39 Studies in mice with sickle cell disease have found that deficiencies in P-selectin result in impaired leukocyte recruitment to the vessel wall, subsequently protecting the mice from vaso-occlusion. 40 Crizanlizumab-tmca is hypothesized to reduce the risk of vaso-occlusion and sickle cell pain crisis by blocking the effects of P-selectin. 41
The efficacy of Crizanlizumab-tmca was evaluated through a phase 2, randomized, multicenter, placebo-controlled, double blind study in patients 16 to 65 years of age with sickle cell disease including HbSS, HbSC, HbSβ+-thal and HbSβ0-thal. 41 Patients had to experience between 2 to 10 pain crises in the past twelve months to be included in the study. Female participants of childbearing potential had to be compliant with birth control methods for the duration of the study and through 4 weeks after completion. 41 Female patients of child bearing age also required a negative serum pregnancy test at screening, prior to randomization, and prior to dosing on day 1 of treatment. 41
Randomized patients were assigned in a 1:1:1 ratio to receive low dose crizanlizumab at 2.5 mg per kilogram of body weight, high dose crizanlizumab at 5 mg per kilogram of body weight, or placebo. Patients received 2 loading doses of crizanlizumab or placebo at 2 weeks apart, followed by a maintenance dose every 4 weeks, up to 50 weeks. The loading and maintenance dosing schedule amounted to a total of 14 doses over a 52-week period. All doses were administered intravenously over 30 minutes. 41
Fewer pain crisis occurred in patients receiving high dose crizanlizumab at a median rate of pain crisis per year of 1.63 (0.00 – 3.97); P = 0.01, compared to the low dose crizanlizumab group 2.01 (1.00 – 3.98); P = 0.18 and the placebo group 2.98 (1.25 – 5.87). Twenty-four patients in the high dose crizanlizumab group experienced zero pain crisis during the trial compared to 12 patients in the low dose crizanlizumab group and 11 patients in the placebo group. 41
The subgroup analysis showed that patients with concomitant hydroxyurea use had a highest median rate of pain crisis per year in the placebo group, followed by the high dose crizanlizumab group, and lastly the low dose crizanlizumab group. 41 Notably, among patients without concomitant hydroxyurea use, the low dose crizanlizumab group had the highest median rate of pain crisis per year, followed by the placebo group, and lastly the high dose crizanlizumab group. 41 The subgroup analysis of patients with 2 to 4 pain crises in the previous year and 5 to 10 pain crises in the previous year, both showed that patients in the high dose crizanlizumab group had the lowest median rate of crisis per year. 41 Patients in the high dose crizanlizumab group also had the lowest annual rate of days hospitalized, and the longest time to first and second sickle cell related pain crisis. 41
Thirty-three percent of the patients in the low dose crizanlizumab group reported at least 1 serious adverse event consisting of pyrexia, influenza, and pneumonia, compared to 26% of patients in the high dose crizanlizumab group, and 27% of patients in the placebo group. 41 The number of patients experiencing any adverse event was similar among the groups at 86% in the high dose crizanlizumab group, 88% in the low dose crizanlizumab group, and surprisingly 89% in the placebo group. 41 The most commonly reported adverse events in the crizanlizumab groups included headache, back pain, nausea, arthralgia, and pain in the extremities. 41 The main limitation to using crizanlizumab is the high monthly cost of the drug. 42
Discussion
Current evidence suggests using hydroxyurea as the mainstay for therapy for most patients with HbSS and HbSβ0-thal. 42 The updated American Society of Hematology (ASH) 2019 and 2020 guidelines focus specifically on cardiopulmonary and kidney disease, cerebrovascular disease, transfusion support, management of acute and chronic pain with no specific mention of the newer agents.43-46 Despite its benefit, hydroxyurea has some major drawbacks. Notably, hydroxyurea is effective in some genotypes – HbSS or HbSβ+-thal or HbSβ0-thal.23,42 Patients who are less adherent to hydroxyurea are more likely to have a higher reported number of VOCs. 42 The study conducted by Niiharo for L-glutamine was mainly done in patients with genotype SS however, there might be some benefit seen in patients who are unable to tolerate hydroxyurea with genotype SS and or patients with genotype SC. 29 However, this study did not evaluate any patients with the SC genotype. 29
Voxelotor and crizanlizumab are 2 relatively new agents that are currently approved to be given with or without hydroxyurea. Currently, voxelotor seems to confer benefit in patients with hypoplastic anemia in sickle cell disease and those who are unable to tolerate or are unwilling to undergo multiple blood transfusions. Thus far, only some insurance companies are covering for voxelotor in patients who have not seen benefit with erythropoietin stimulating agents (ESAs). Encouragingly, patients are more compliant with voxelotor as they prefer the oral agent to biweekly injections of ESAs. Although voxelotor is still less utilized in this patient population, the results of the HOPE study showed that patients with higher hemoglobin levels had significantly reduced numbers of VOCs.33,34
Crizanlizumab is an option in patients experiencing VOCs but its use is limited by the high monthly cost. Crizanlizumab is considered a specialty drug, therefore the process of obtaining it for patients without commercial insurance is difficult. Most hospital pharmacies have been reluctant to carry the medication, therefore it must be ordered at a specialty pharmacy and then sent to the hospital pharmacy for mixing. Once the mixing is done, the medication is then delivered to the outpatient infusion center to be administered to the patient once a month. In comparison to hydroxyurea, crizanlizumab has a lengthy prior authorization process taking as long as 3 months to get approved with little incentive for hospital pharmacies to add it to their formulary inventory. 42
Therefore, the following factors need to be taken into consideration when selecting an agent to reduce VOCs. First and foremost, the cost of the medication and the type of insurance will determine the agent selected. Both voxelotor and crizanlizumab have annual costs of $100,000 compared to an annual cost of $1,000 for hydroxyurea and $20,000 for L-glutamine.
42
With hydroxyurea the incidence of VOC is reduced by half but only 30 percent of patients are compliant with taking the medication.
42
Some patients have no employment or are on public insurance plans therefore making it harder to justify the 2 new expensive medications.
42
The other factor is the patient’s genotype since patients with genotype HbSS have more painful crises and could potentially benefit more from hydroxyurea and crizanlizumab. Another factor that needs to be taken into account is medication dose adjustments. Patients on hydroxyurea have to be dose adjusted if the creatinine clearance is

Prevention
Patients with SCD should be aware of factors that may precipitate a VOC such as infections, low oxygen tension, dehydration, acidosis, physical or psychological stress, alcohol use, pregnancy, cold weather, and concomitant conditions such as sarcoidosis, diabetes mellitus, and herpes.12,13,16 It is essential for patients to be counseled on nonpharmacologic strategies to prevent VOCs such as drinking adequate amounts of fluid to avoid dehydration, avoiding mountain climbing and flying in unpressurized cabins above 10,000 feet, avoiding cold weather, avoiding exercising especially in the heat to the point of dehydration, and avoiding drugs that may lead to acidosis. 15
Conclusion
Needless to say, for many patients with sickle cell disease, public health insurance is a major limiting factor in their access to the newer agents. Currently, many patients with VOCs will still benefit from taking hydroxyurea given the long-term efficacy data and low cost associated with the medication when compared to the newer agents including L-glutamine, voxelotor and crizanlizumab. However, barring access, patients with sickle cell disease now have more than one pharmacologic option to manage and prevent VOCs with many more agents being developed down the pipeline.
Footnotes
Authors’ Note
Dr Divita Singh is now affiliated with Temple University School of Pharmacy, Department of Pharmacy Practice, Philadelphia, PA, USA.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.