
Abstract
Traumatic brain injury (TBI) is a neurological disease that seriously endangers human life and has a poor prognosis. In particular, neuroinflammation during secondary injury after TBI affects the course of TBI, and interleukin-33 (IL-33) plays an important regulatory role in neuroinflammation after TBI. Meanwhile, the Yes-associated protein (YAP) can influence the prognosis after TBI. In this study, we explored whether the upregulation of YAP in astrocytes can enhance the protective effect of IL-33 against neuroinflammation after TBI. In the current study, the markers of microglial proinflammatory/anti-inflammatory responses both in vivo and in vitro were assessed after the administration of exogenous IL-33. Adeno-associated virus targeting astrocytes in vivo and lentivirus transfecting astrocytes in vitro were used to overexpress YAP, and the expression and localization of proteins were evaluated by Western blotting and immunofluorescence staining. Chromatin immunoprecipitation-quantitative Polymerase Chain Reaction (qPCR) assays were performed to confirm that YAP transcriptionally regulates the IL33 gene by binding directly to its promoter region. Astegolimab was administered to block Growth Stimulation Express Gene 2 Protein (ST2) receptors in vivo and in vitro. Morris water maze and Y-maze tests were employed to assess cognitive function after TBI. The results demonstrated that the expression levels of both YAP and IL-33 were significantly decreased during the early phase of TBI. Concurrently, the anti-inflammatory marker CD206 in microglia was also markedly reduced in the acute stage post-TBI. Importantly, YAP was found to enhance IL-33 secretion by binding to its gene promoter, thereby activating the IL-33/ST2 signaling pathway. This activation promoted anti-inflammatory responses in microglia, which were mediated through the NF-κB signaling pathway, and ultimately led to improved cognitive function. These beneficial effects were effectively reversed by the administration of astegolimab, confirming the specificity of the YAP/IL-33/ST2 mechanism. Above all, we found that YAP produced by astrocytes regulates microglial anti-inflammatory responses through the IL-33/ST2 pathway, thereby improving cognitive function after TBI.
Keywords
Introduction
Traumatic brain injury (TBI) has the highest incidence among neurological disorders, which leads to an immense health burden. 1 The pathophysiology of TBI involves two main phases: primary brain injury, which refers to the immediate mechanical damage, and secondary brain injury, which encompasses prolonged processes, such as neuroinflammation, that contribute to neurological deficits.2–4 Neuroinflammation mediated by astrocytes and microglia has an important effect on the prognosis of TBI.5,6
Interleukin-33 (IL-33), a member of the IL-1 superfamily, plays a crucial role in innate immunity and inflammatory responses.7,8 It could bind its orphan receptor ST2 on microglia, and this ligand–receptor interaction enables communication with astrocytes; upon receiving IL-33 signals released by astrocytes, microglia become activated and exhibit enhanced phagocytic function.9,10 Other researchers also found the protective effect of IL-33 against neuroinflammation following central nervous system injury, such as cerebral ischaemia, 11 TBI, 12 and brain Toxoplasma gondii infection. 13 Besides, chronic inflammatory activation is regarded as a contributing factor to cognitive dysfunction.5,14 As we know, the hippocampus plays a significant part in learning and spital memory. 15 Previous research has found that IL-33 can influence homeostatic synaptic plasticity in the adult hippocampus and decrease the induction of anti-inflammatory genes of microglia,16,17 But few precursor factors that affect the function of IL-33 have been identified.
Yes-associated protein (YAP), as the key transcriptional regulator downstream of the Hippo signaling pathway, plays an important role in organ growth, tissue renewal, and regeneration. 18 Pathologically, the impaired activation of YAP within the Hippo pathway can lead to deficient tissue repair and exacerbate secondary injury, thereby adversely affecting outcomes after TBI.19,20 The YAP protein has been found to play a crucial role in regulating the promoter of the IL-33 cytokine following nuclear localization, thereby regulating cardiac fibroblast activation and the fibroinflammatory response. 21 However, the role of YAP in regulating neuroinflammation after TBI remains poorly understood.
Our current study demonstrates that the expression of YAP and IL-33 in astrocytes decreases during the early phase after TBI, accompanied by a reduction in the anti-inflammatory response of microglia. We further confirmed that the exogenous administration of IL-33 effectively enhances the anti-inflammatory response of microglia and improves cognitive function. Through lentiviral and adeno-associated virus (AAV)-mediated overexpression of YAP in astrocytes and mice, respectively, we observed a significant upregulation and activation of IL-33. Using astrocyte–microglia co-culture systems, we found that YAP overexpression in astrocytes influences the anti-inflammatory response of microglia and improves cognitive outcomes after TBI. We show that YAP, as a downstream transcription factor of the Hippo signaling pathway, is significantly reduced in the nuclei of astrocytes during the early post-TBI phase. Additionally, chromatin immunoprecipitation (CHIP)-qPCR experiments confirmed that YAP binds to the promoter region of the IL33 gene in astrocytes. Treatment with astegolimab, a specific blocker of the IL-33 receptor ST2, reversed the enhanced anti-inflammatory response in microglia induced by YAP overexpression in astrocytes and impaired cognitive recovery. Furthermore, we demonstrated that IL-33/ST2 signaling influences the NF-κB pathway in microglia, which plays a critical role in regulating their anti-inflammatory response. These findings indicate that YAP in astrocytes participates in early neuroinflammatory responses following TBI through IL-33/ST2 signaling. By modulating microglial function through multiple mechanisms, targeting the YAP/IL-33 axis may represent a promising therapeutic strategy for mitigating neuroinflammation in the early stage after TBI.
Materials and Methods
Human brain samples
Human brain tissues were obtained from Nanjing Jinling Hospital, Affiliated Hospital of Medical School, Nanjing University, Nanjing, China. Six TBI tissues and six noncontusive tissues (control) were included. Detailed information of the six TBI tissues and six control tissues were shown in Table 1. Six control tissues were obtained from the surgical approach during surgery of meningioma or glioma. Human brain tissue and blood were collected using a protocol approved by the Ethics Committee of Jinling Hospital (approval No. 2024DZGJJ-190).
Demographic and Clinical Characteristics of TBI and Control (Con) Patients

Animal preparation and experimental design
All procedures involving animals conformed to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health and were approved by the Institutional Animal Care and Use Committee of Jinling Hospital.
Adult male C57/BL6 mice (20–25 g) were purchased from the Model Animal Research Center of Nanjing University (Nanjing, China). All mice were housed in cages at a constant temperature (26 ± 2°C), air humidity of 50% ± 10%, and a 12-h light/dark circle, with food and water available ad libitum. The experimental design is shown in Supplementary Fig. S1A.
Cell culture and experimental design
Mouse astrocyte type I cells (C8-D1A) and BV-2 microglial cells (BV-2) were purchased from Zhong Qiao Xin Zhou Biotechnology Co., Ltd. (Shanghai, China). The cells were maintained at 37°C in serum-containing nutrient medium consisting of 89% high-glucose Dulbecco’s modified Eagle medium (Cat. No. 11995065, Gibco BRL, Grand Island, NY, USA), 10% fetal bovine serum (Cat. No. 10099141, Gibco BRL), and 1% penicillin–streptomycin (all from Gibco BRL). C8-D1A and BV-2 cells were then passaged to 6-well plates or co-culture chambers for further experiments. All the medium were then incubated in humidified air of 5% CO2 at a temperature of 37°C. The experimental design is shown in Supplementary Fig. S1A.
TBI model in vivo
We used a modified version of the weight drop model. In the experiment, according to the mortality of mice, the model we used should be considered as severe brain injury. 22 As described previously, mice were anesthetized by 2% isoflurane and 100% oxygen, which was maintained with 1% isoflurane, and after the skull was exposed, the weight drop device was dropped 2.0 mm posterior and lateral to the bregma.23,24 TBI was induced by a weight of 100 g falling from a height of 5 cm. The sham group of mice underwent scalp dissection only, but no injury was caused. During surgery, the body temperature of the mice was maintained at 37°C with a homeothermic heating pad. We returned the mice to their cages 20 min after surgery.
TBI model in vitro
When the microglia and astrocytes were cultured alone or in the co-culture system, the in vitro TBI model was performed using the corresponding culture medium mentioned above. The TBI model was performed based on the previous studies with minor modifications.25–28 Briefly, we induced the injury with a scalpel cut 20 times through the astrocyte and microglia culture medium or co-culture chamber, 10 times perpendicularly in either direction, approximately 2-mm apart.
Lentiviral transfection
The Yap1 gene (NCBI reference sequence ID, NM_001171147) overexpression lentivirus was constructed and purchased from Hanbio Biotechnology (Shanghai, China). The astrocytes were incubated with the lentivirus for 48 h before being used for further experiments (cell lysis or co-culture).
IL-33 and anti-IL-33 administration
Based on the previously described method, drug administration was carried out with slight alterations.29,30 Briefly, in vitro cultures were treated with recombinant mouse IL-33 protein (HY-P7218, MedChemExpress) and an anti-IL-33 antibody (AF3626, R&D Systems) at a concentration of 300 ng/mL in culture medium for 24 hours. The IL-33 protein (40 ng/mouse) and anti-IL-33 (40 ng/mouse) were administered via intracerebroventricular injection with a total volume of 5 µL at 30 min after TBI induction in vivo. An equal volume of PBS was used as a negative control. The details of intracerebroventricular administration were as mentioned below.
Intracerebroventricular injection of AAV and astegolimab
AAV (targeting astrocytes) purchased from Gene Chem Co. (Shanghai, China) was injected, in order to overexpress YAP in astrocytes. AAV was administered into the right ventricle 14 days before the induction of TBI. Under sterile conditions, the injection site (1.5 mm lateral and 0.8 mm posterior to the bregma, at a depth of 2.4 mm) was exposed for the administration of AAV. The lateral ventricle received a vertical injection of AAV, with a total volume of 5 µL and a flow rate of 0.5 µL/min. Next, the microsyringe was allowed to sit for a duration of 5 min in order to avoid any reverse flow. The negative control mice received the same quantities of AAV containing mock injected into the lateral ventricles. Astegolimab (HY-P99444, MedChemExpress) was prepared at 0.8 mg/ml in PBS. Astegolimab was administered via intracerebroventricular injection with a total volume of 5 µL at 30 min before TBI induction. The equal volume of PBS was used as a negative control.
Western blotting
Based on the previous method, 24 the mice used for tissue extraction in this experiment were all injected with ice-cold normal saline under deep anesthesia, and the hippocampal tissue of the lesion and the injured cortex were respectively harvested and stored at −80°C. Brain tissues and cells were lysed in radio immunoprecipitation assay buffer containing 1% phenylmethanesulfonyl fluoride (ST506, Beyotime) and 1% phosphatase and protease inhibitor cocktail (P5726, Sigma). To obtain the nucleoprotein, we used a nuclear and cytoplasmic protein extraction kit (P0028, Beyotime). After separation by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, the protein bands were electrotransferred to a polyvinylidene difluoride membrane. After blocking the membrane with 5% skim milk at room temperature for 2 h, we incubated the membrane with primary antibodies against β-actin (1:4000, GB15003, Servicebio), YAP (1:1500, 14074, Cell Signaling Technology), P-YAP (1:1500, 4911, Cell Signaling Technology), IL-33 (1:1000, AF3626, R&D), ST2 (1:2000, 11920-1-AP, Proteintech), glial fibrillary acidic protein (GFAP, 1:4000, 2118, Cell Signaling Technology), histone 3 (H3,1:3000, 17168-1-AP, Proteintech), CD206 (1:1500, ab64693, Abcam), CD16 (1:1500, 16559-1-AP, Proteintech), C3 (1:1500, 21337-1-AP, Proteintech), S100A10 (1:1500, 11250-1-AP, Proteintech), NF-κB (1:1500, 8242S, Cell Signaling Technology), and phospho-NF-κB(1:1500, 3033S, Cell Signaling Technology). Then, the membranes were washed three times (10 min each time) with Tris-buffered saline (TBS) containing Tween 20 before incubation with the species-matched secondary antibody (1:5000) at room temperature for 2 h. Following incubation, the membranes were washed three times (15 min each time) with TBS containing Tween 20. Finally, the protein bands were visualized via an enhanced chemiluminescence system, and the relative density was analyzed using ImageJ software. All groups included n = 6 biologically independent replicates.
Immunofluorescence staining
Immunofluorescence staining of the mice brain sections was performed as previously described. 29 In brief, the mouse brain sections were incubated with primary antibodies against YAP (1:200, 14074, Cell Signaling Technology, Danvers, MA, USA), IL-33 (1:200, AF3626, R&D), GFAP (1:200, 60190-1-Ig, Proteintech), IBA-1 (1:200, Servicebio GB12105), CD206 (1:1500, ab64693, Abcam), and CD16 (1:1500, 16559-1-AP, Proteintech) overnight at 4°C. After washing with phosphate-buffered saline (PBS) containing Tween 20 (8 min each time), the brain sections were incubated with the appropriate fluorescently labeled goat anti-rabbit IgG antibodies (1:200, SA00009-2, Proteintech) or goat anti-mouse IgG antibody (1:200, SA00013-1; Proteintech) or donkey anti-goat IgG antibodies (1:200, A24431, Abbkine) at room temperature for 2 h. Following incubation with 4’,6-diamidino-2-phenylindole (DAPI) solution (C1005, Beyotime) at room temperature for 5 min, the brain slices were sealed with anti-fluorescence quenching-mounting solution. Finally, the brain slices were imaged using a fluorescence microscope. The mean fluorescence intensity was quantified using ImageJ. All groups included n = 3 biologically independent replicates.
Y-maze test
We employed the Y-maze test as previously described to evaluate the short-term spatial memory in mice. 29 The apparatus consists of three symmetrical arms (each measuring 30 cm in length and 8 cm in width) at an angle of 120° from each other. When the test began, the mice were placed in one arm of the apparatus and allowed to explore the environment for 8 min. Consecutive participation into all three arms was considered as a correct alteration. The alternation percentage was calculated as the ratio of correct alternations to maximum alternations. The total number and the sequence of arm entries were video recorded. All groups included n = 6 biologically independent replicates.
Morris water maze test
In this study, we performed the Morris water maze (MWM) test, as previously described. 31 In a nutshell, the MWM assay was used to assess learning ability and spatial memory function in mice. The experiment consisted of two tests, including 6 consecutive days of the place navigation test and 1 day after that of the spatial probe test. Starting on the 24th day after TBI and ending on the 29th day, mice were sent randomly from one of the three quadrants (the first, the second, or the third) without the platform of the pool and allowed to seek the hidden platform for 60 sec. On the 30th day, the platform was withdrawn, and the spatial probe test was conducted to record the quadrant time and platform crossovers of each mouse. All groups included n = 6 biologically independent replicates.
Chromatin immunoprecipitation assay
Following the manufacturer’s instructions, the ChIP assay was conducted using the Magna ChIP® A/G Chromatin Immunoprecipitation Kit (17-10085, Millipore, Bedford, MA, USA). In brief, the proteins and DNA were crosslinked using 1% formaldehyde, and the cell lysates were sonicated to acquire DNA fragments. The DNA fragments were then subjected to immunoprecipitation with primary antibody against YAP (1:50, 14074, Cell Signaling Technology) or negative control IgG (A7016, Beyotime, Nantong, China). Purified DNA was then analyzed via qRT-PCR with the SYBR Green qPCR Master Mix (Q711, Vazyme, Nanjing, China). Finally, the relative enrichment was calculated after normalizing the results to the input values. The primers used are listed in Supplementary Table S2.
Statistical analysis
All data were analyzed using GraphPad Prism 9.0, and the values of each group are presented as mean ± standard deviation. Statistical differences among three or more groups were analyzed by one-way analysis of variance (ANOVA). If a significant difference was determined by ANOVA, Tukey’s post hoc multiple comparison test was employed. Student’s t test was conducted when only two groups were compared. We considered statistical differences to be significant in cases where the p value was <0.05.
Results
Decreased IL-33 and YAP expression in patients with TBI
All brain tissues were obtained from Sample Bank of Nanjing Jinling Hospital, Affiliated Hospital of Medical School, Nanjing University, Nanjing, China. The patient specimen information was displayed in Table 1. According to the relevant clinical research and basic research,12,32 IL-33 is known to be involved in the pathological process and influences the progression and prognosis of TBI. We first used Western blotting to analyze IL-33 expression in human brain tissue after TBI. The results showed that the expression level of IL-33 in the TBI group was lower than that in the control group (Fig. 1A, B). Next, we detected the co-localization of astrocytes and IL-33 after TBI by immunofluorescence staining (Fig. 1F, G). The results demonstrated that the intensity of IL-33 was lower than that of the control group after TBI. Based on a previous study,19–21 we hypothesized that YAP could affect the cognitive function after TBI by regulating the expression of IL-33. For the reasons mentioned above, we analyzed the YAP expression level in human brain tissue after TBI. As expected, the YAP protein expression level (Fig. 1A, D) and the immunofluorescence staining intensity (Fig. 1H, I) decreased significantly in the TBI group compared with the control group. Collectively, these results indicate that both the IL-33 and YAP expression levels are substantially decreased in human brain tissues following TBI.
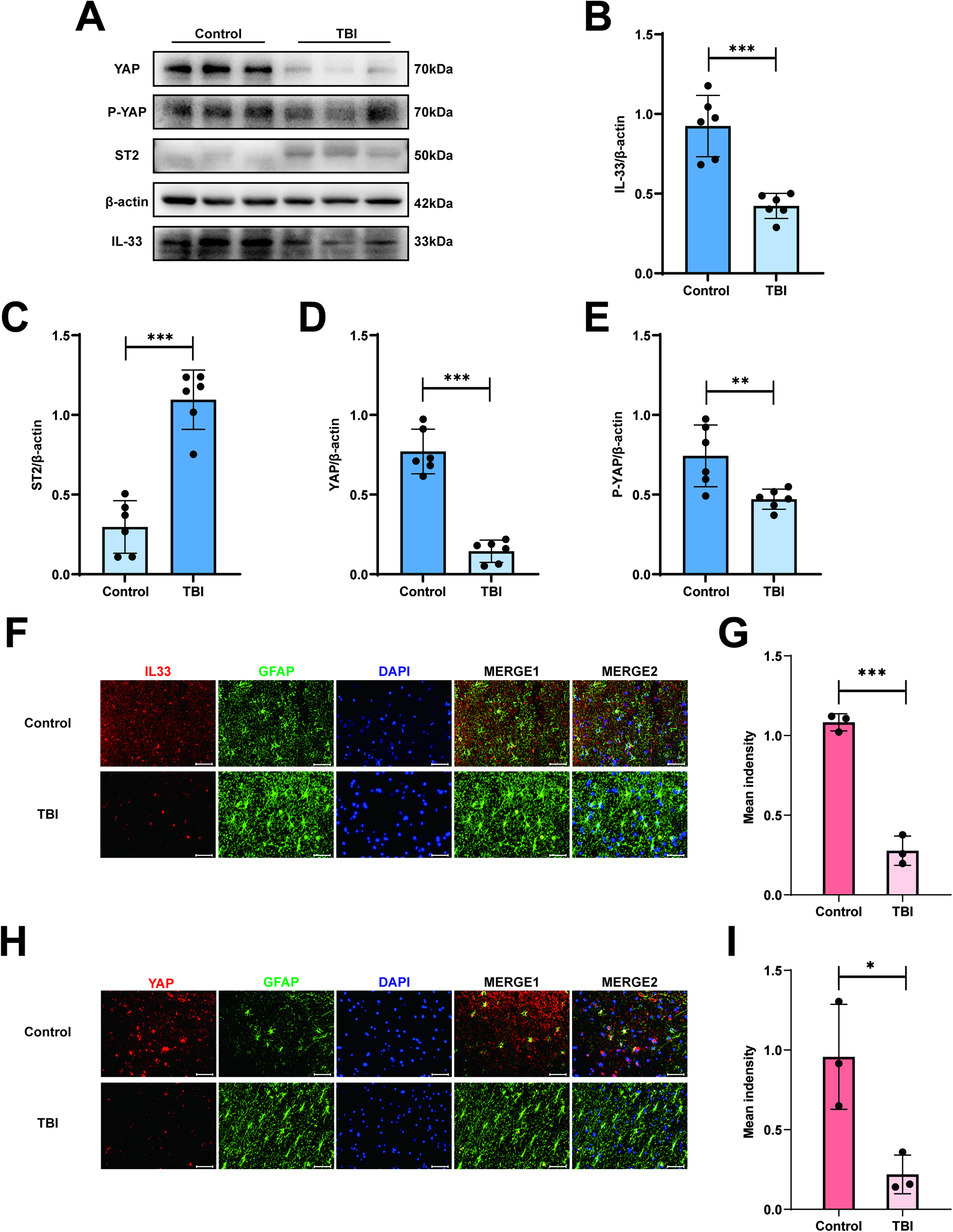
Decreased IL-33 and YAP expression in patients with TBI. (
IL-33 expression decreased in the early stage after TBI in experimental model
In our study, we succeeded in establishing the model of experimental TBI in mice by following a previous study. 24 The mice were divided into two groups at random: the SHAM group and the TBI group. As shown in Supplementary Fig. S1B, compared with the SHAM group, there were obvious blood and injury lesions in the modeling areas in the TBI 3d group. We first used Western blotting to analyze the time-course change of IL-33 in astrocytes of the mouse brain after TBI. As depicted in Figure 2A–D, the expression level of IL-33 in the cortex and hippocampus decreased during the early phase after TBI, reaching their lowest point in the TBI 3d group. The next step was to determine whether there are similar changes in vitro. The injury was induced with a scalpel cut 20 times through the medium-cultured astrocytes, 10 times perpendicularly in either direction, approximately 2-mm apart. After scratching the astrocytes, we saw the decreased level in IL-33 expression, especially in the TBI 24 h group by Western blotting. (Fig. 2E, F). As expected, IL-33 co-localized with the astrocyte marker GFAP, and the immunofluorescence intensity of IL-33 decreased in the TBI 3d group compared with the Sham group in the cortex (Fig. 2G, H) and hippocampus (Fig. 2I, J) of mice brain tissue. All of the above results indicated that IL-33 expression decreased in the early stage following TBI in both in vivo and in vitro experimental models.
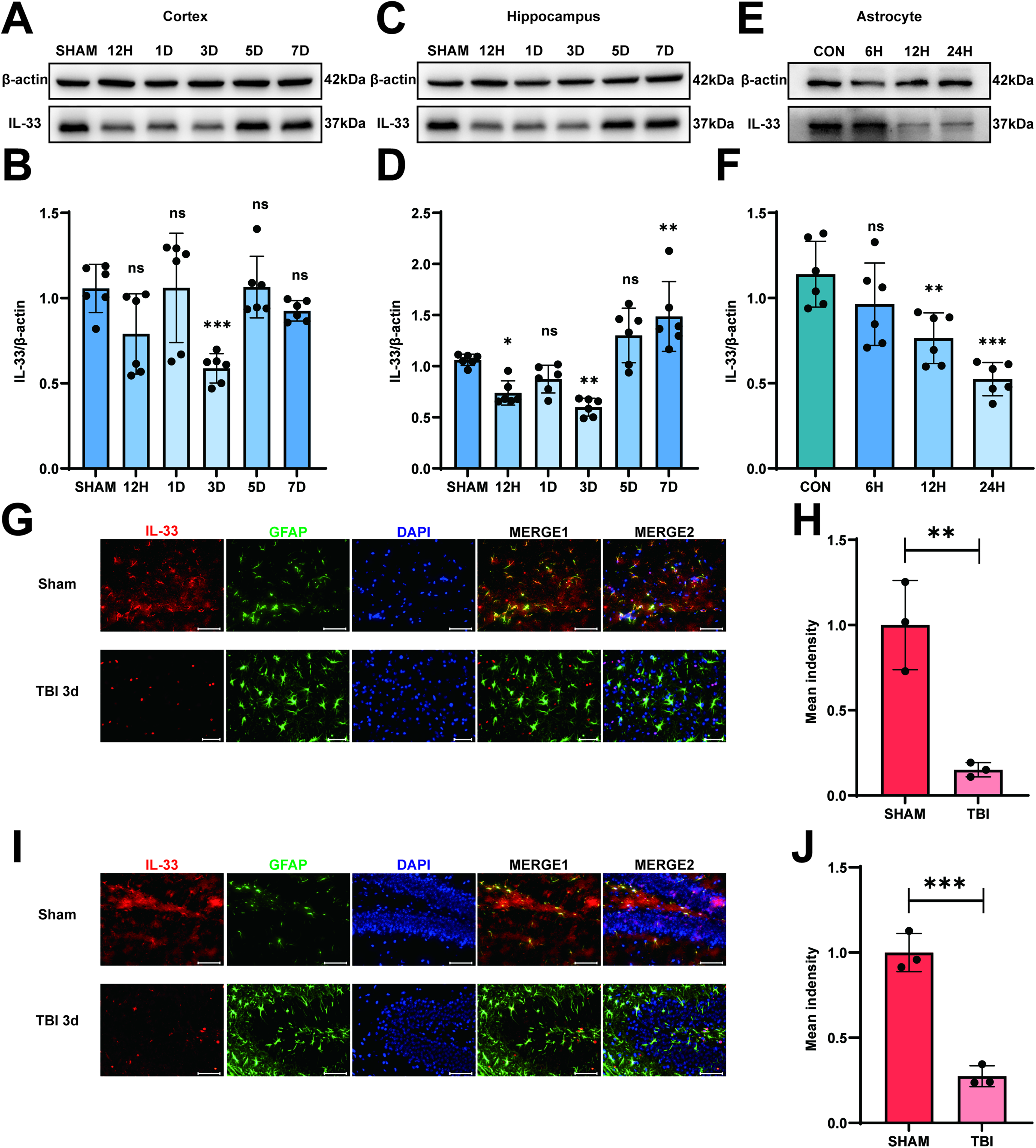
IL-33 expression decreased in the early stage after TBI in experimental model both in vivo and in vitro. (
Administration of IL-33 regulated anti-inflammatory responses of microglia, contributing to the improvement of cognitive function after TBI
The mice were divided into five groups at random: SHAM, TBI, TBI + PBS, TBI + IL-33 and TBI + Anti-IL-33 groups. Based on previous experiments, we selected day 3 post-TBI as the time point for conducting further experimental research. To verify whether neuroinflammation in the early stage of TBI was related to changes in IL-33 expression, we used CD16 and CD206, which are receptor proteins on the surface of microglia, as indicators of microglial proinflammatory/anti-inflammatory responses.33–35 Exogenous IL-33 was administered via intracerebroventricular injection to elevate brain IL-33 levels. As shown in Figure 3A–C, the cortex CD206 expression levels decreased in the TBI group compared with those in the SHAM group, and there was no significant difference between the TBI and TBI + PBS groups. While the CD206 expression level increased in the TBI + IL-33 group compared with that in the TBI group, there was no significant difference between the TBI and TBI + anti-IL-33 groups. However, the CD16 expression level had no significant difference between the TBI + IL-33 and TBI groups. CD206 or CD16 expression levels showed the same trend in the hippocampus respectively (Fig. 3D,F). Consistent with the in vivo results, the scratch-induced in vitro model of microglial activation showed analogous changes in CD206 and CD16 expression (Fig. 3G–I). These findings suggest that elevated IL-33 promotes anti-inflammatory responses in microglia. We next detected whether CD206 co-localized with the microglial marker IBA-1 and the immunofluorescence intensity change. As shown in Figure 4A,B, the immunofluorescence staining intensity of CD206 showed the same trend as the CD206 expression level in the analysis of Western blotting in the hippocampus. The corresponding immunofluorescence staining in the cortex is shown in Supplementary Fig. S2A,B. Not surprisingly, the results of the immunofluorescence colocalization and intensity analysis of CD16 and IBA-1 were consistent with those of Western blotting analysis, both in the hippocampus (Fig. 4C,D) and cortex (Supplementary Fig. S2C,D). The results of the Y-maze test displayed higher spontaneous alternations in the SHAM and TBI + IL-33 groups, indicating better spatial working memory (Fig. 4E). Following the exogenous IL-33 administration in the mice brain, the escape latency after TBI dropped significantly from training day 5 to day 6 (Supplementary Fig. S2E). Furthermore, in the probe test, the SHAM and TBI + IL-33 group mice tended to spend more time in the target quadrant and cross the platform more frequently compared with those in the TBI group (Fig. 4F–H), while no significant difference was observed in the swim speed among the groups of mice (Supplementary Fig. S2F). The data demonstrated that exogenous IL-33 improved cognitive function after TBI.
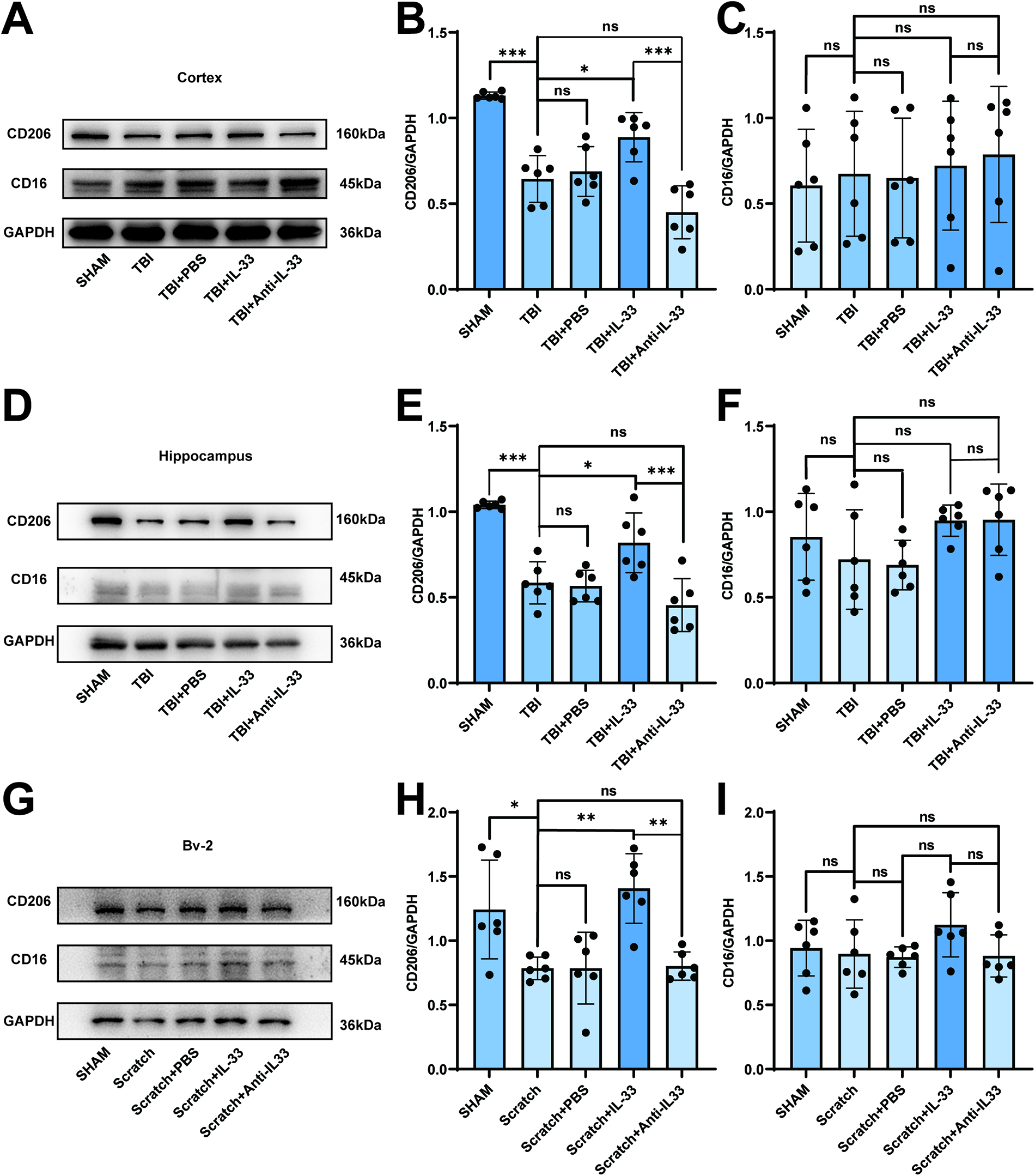
Administration of IL-33 regulates anti-inflammatory responses of microglia after TBI.
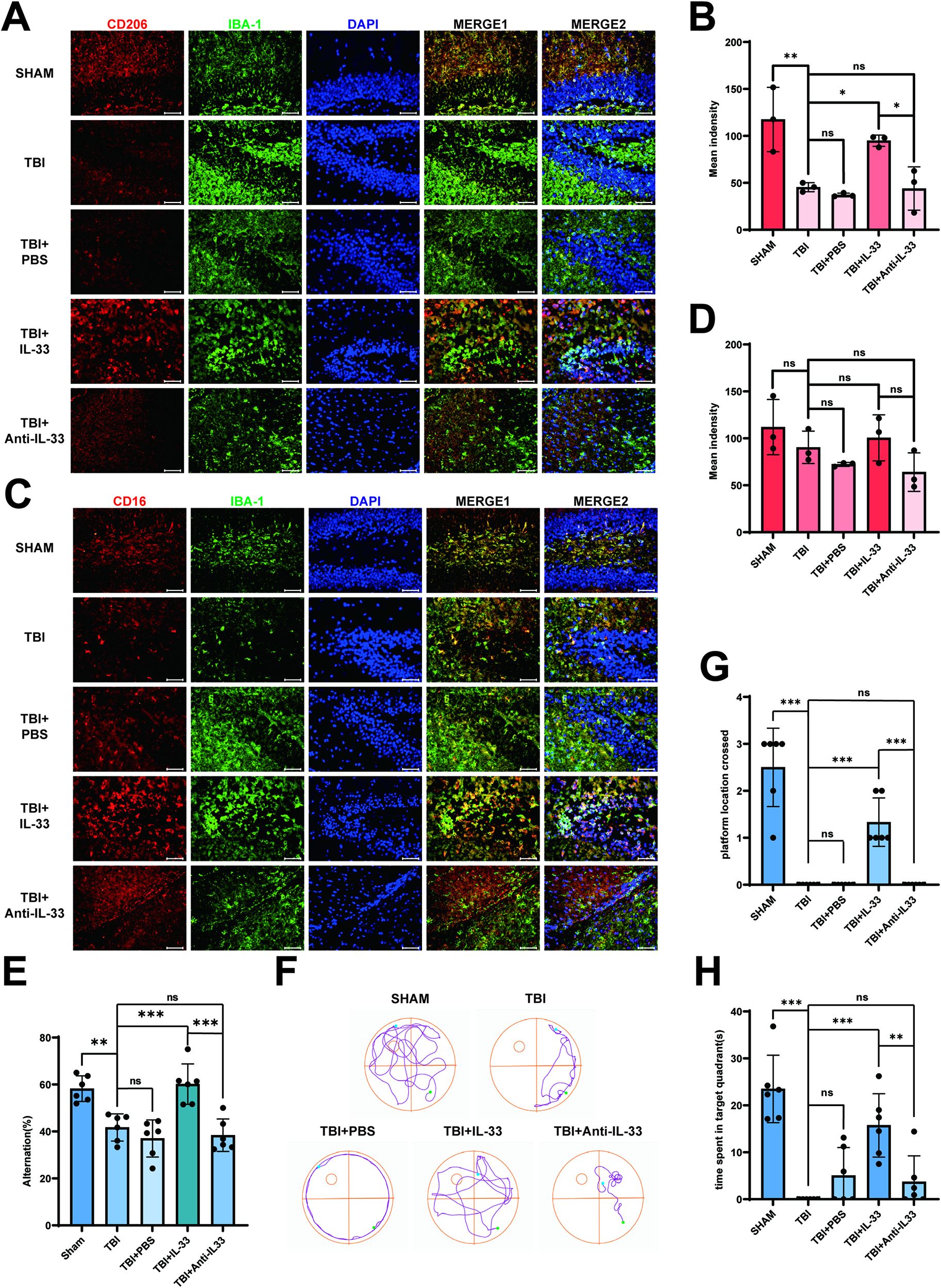
Administration of IL-33 regulates anti-inflammatory responses of microglia after TBI and improves cognitive function after TBI. (
The change in YAP expression level regulates the secretion of IL-33 by astrocytes
To explore the cause of this time-dependent IL-33 expression level, we examined the protein level of YAP in the cortex and hippocampus at 12 h, 24 h, 3 days, 5 days, and 7 days after the induction of experimental TBI. Interestingly, we discovered that the expression of YAP decreased in the cortex (Fig. 5A,B) and hippocampus (Fig. 5C,D) on day 3, which was consistent with the trend of IL-33 expression. We extended the modeling period of the TBI mouse model to 2 and 4 weeks and performed Western blot analysis to examine the protein expression changes of YAP and IL-33. Both YAP and IL-33 protein levels showed a trend of recovery or even upregulation during the longer-term neuroinflammatory phase after TBI (Supplementary Fig. S3A–C). P-YAP is an inactivated form of YAP that cannot be transported into the cell nucleus,18,36 but we found that the P-YAP expression showed no significant change in the cortex and hippocampus (Supplementary Fig. S4A–D). After establishing the experimental model in vitro, we found the expression of YAP protein decreased significantly in the TBI 24 h group (Fig. 5E,F). Immunofluorescence staining revealed YAP co-localization with the astrocyte marker GFAP, along with a reduction in YAP intensity in the TBI 3d group compared with the Sham group in both the cortex and hippocampus (Fig. 5G–J). As previous studies have shown that YAP acts as a co-transcription factor downstream of the Hippo signaling pathway through nuclear translocation,18,36 we specifically measured the nuclear YAP expression level after TBI, and it turns out that the nuclear YAP expression level showed a significant decrease in the TBI 3d group both in the cortex and in hippocampus (Fig. 6A–D), which showed the same trend as the total expression level measured previously (Fig. 5A–D). We used Primer Premier 5 software to predict the potential promoter region of the IL-33 gene and designed corresponding primers for experimental validation. Then, we performed ChIP-qPCR to verify the existence of the combination of YAP and the promoter region, and we found that in the scratch-induced in vitro TBI model, YAP combined significantly fewer fragments of the promoter region (Supplementary Fig. S5A,B). These results implied that the decreased level of YAP may cause astrocytes to secrete less IL-33 after TBI.
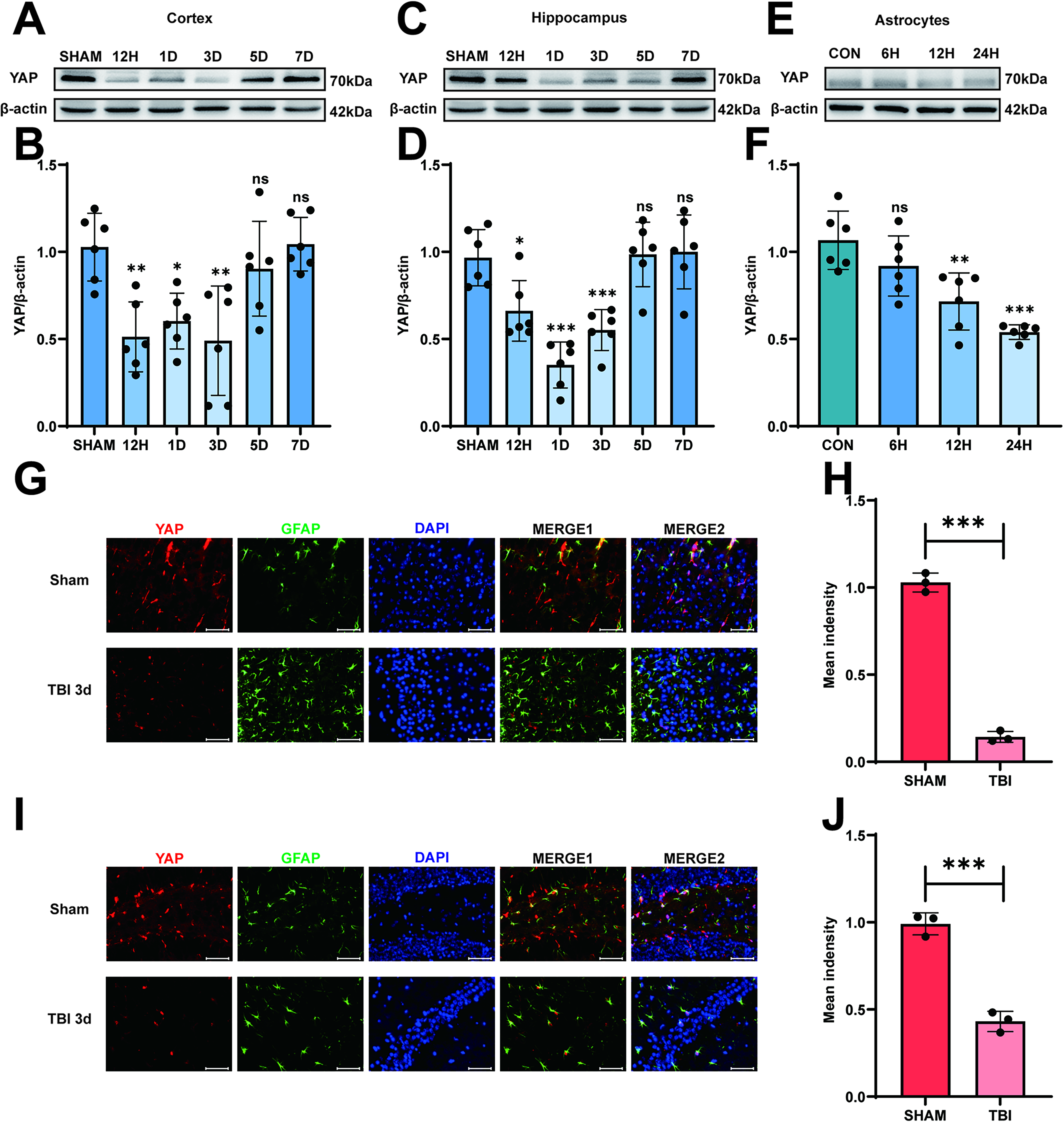
YAP expression decreased in the early stage after TBI in experimental model both in vivo and in vitro. (
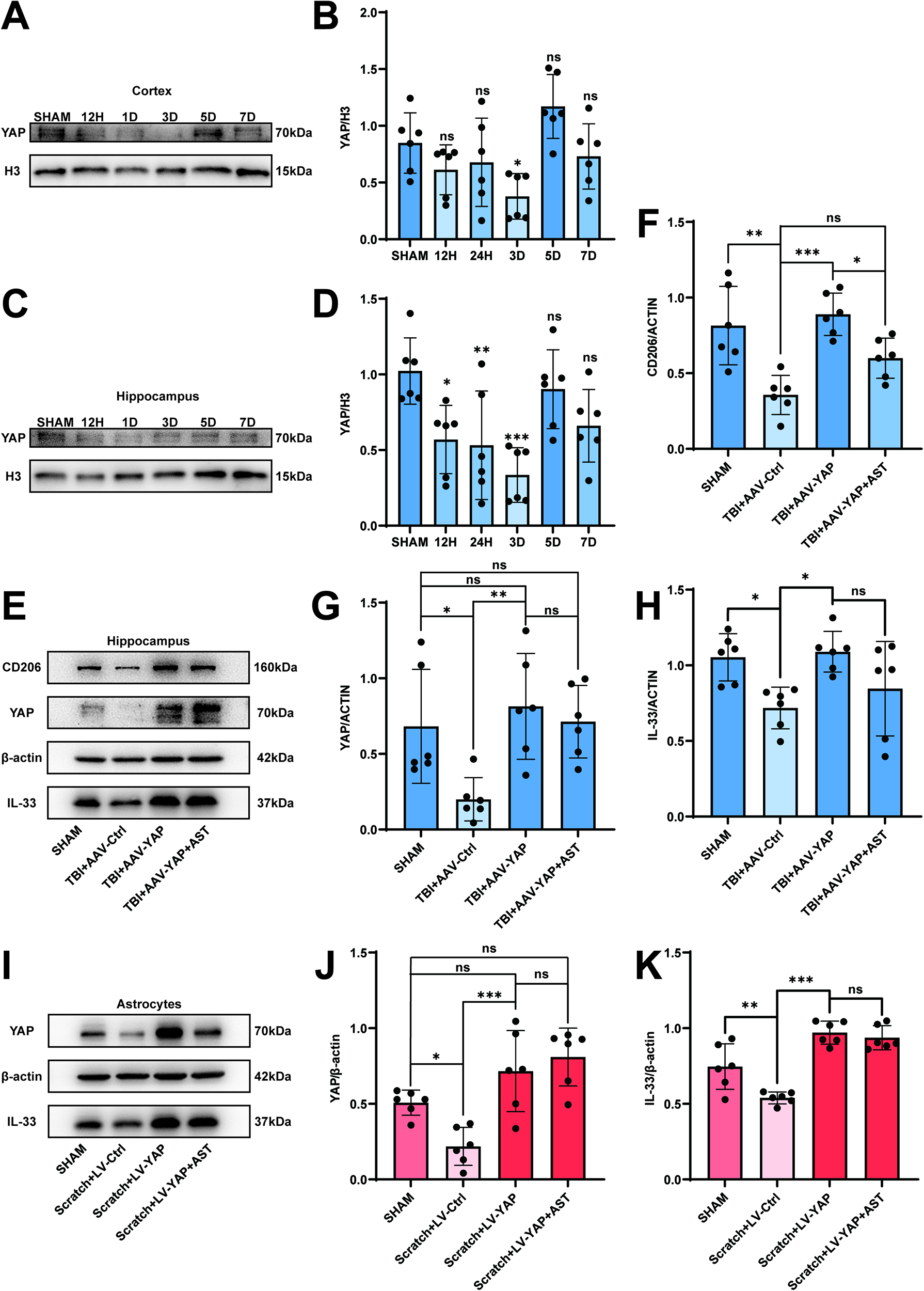
Overexpression of YAP promotes the expression of IL-33 both in vivo and in vitro and decreases the nuclear YAP expression level after TBI. (
Overexpression of YAP promoted anti-inflammatory responses of microglia, leading to improved cognitive function after TBI, while astegolimab reversed the effect of YAP overexpression on the activation of IL-33/ST2 signaling
YAP promotes microglial anti-inflammatory response and cognitive recovery
To investigate whether YAP overexpression in astrocytes modulates microglial polarization in the early stage of TBI, we performed the intracerebroventricular injection of astrocyte-specific AAV to overexpress YAP. Western blotting analysis revealed that hippocampal YAP expression was significantly increased in the TBI + AAV-YAP group compared with the TBI + AAV-Ctrl group (Fig. 6E,G). A similar upregulation trend was observed for IL-33 protein levels (Fig. 6F,H). Consistent with the in vivo findings, in vitro scratch-induced TBI models also showed elevated YAP and IL-33 expression in the Scratch + LV-YAP group compared with the Scratch + LV-Ctrl group (Fig. 6I–K). To assess astrocyte function following overexpression, we performed the Western blotting analysis of the astrocyte marker proteins C3 and S100A10, which are associated with the A1 and A2 phenotypes, respectively.37–39 The results showed no statistically significant differences in the expression levels of these markers, either after viral transfection-mediated YAP overexpression or following treatment with astegolimab (Supplementary Fig. S6A–C).
We further assessed microglial polarization by examining the expression of the anti-inflammatory marker CD206 and the proinflammatory marker CD16. Western blot results demonstrated that CD206 expression was significantly increased in the TBI + AAV-YAP group relative to the TBI group in both the cortex (Fig. 7A–C) and hippocampus (Figs. 6E,F; 7D–F), whereas CD16 levels remained unchanged. Similar results were observed in the co-culture model: microglia cultured with YAP-overexpressing astrocytes exhibited higher CD206 expression without significant changes in CD16 (Fig. 7G–I). Immunofluorescence staining confirmed these trends in the hippocampus (Fig. 8A–D) and cortex (Supplementary Fig. S7A–D).
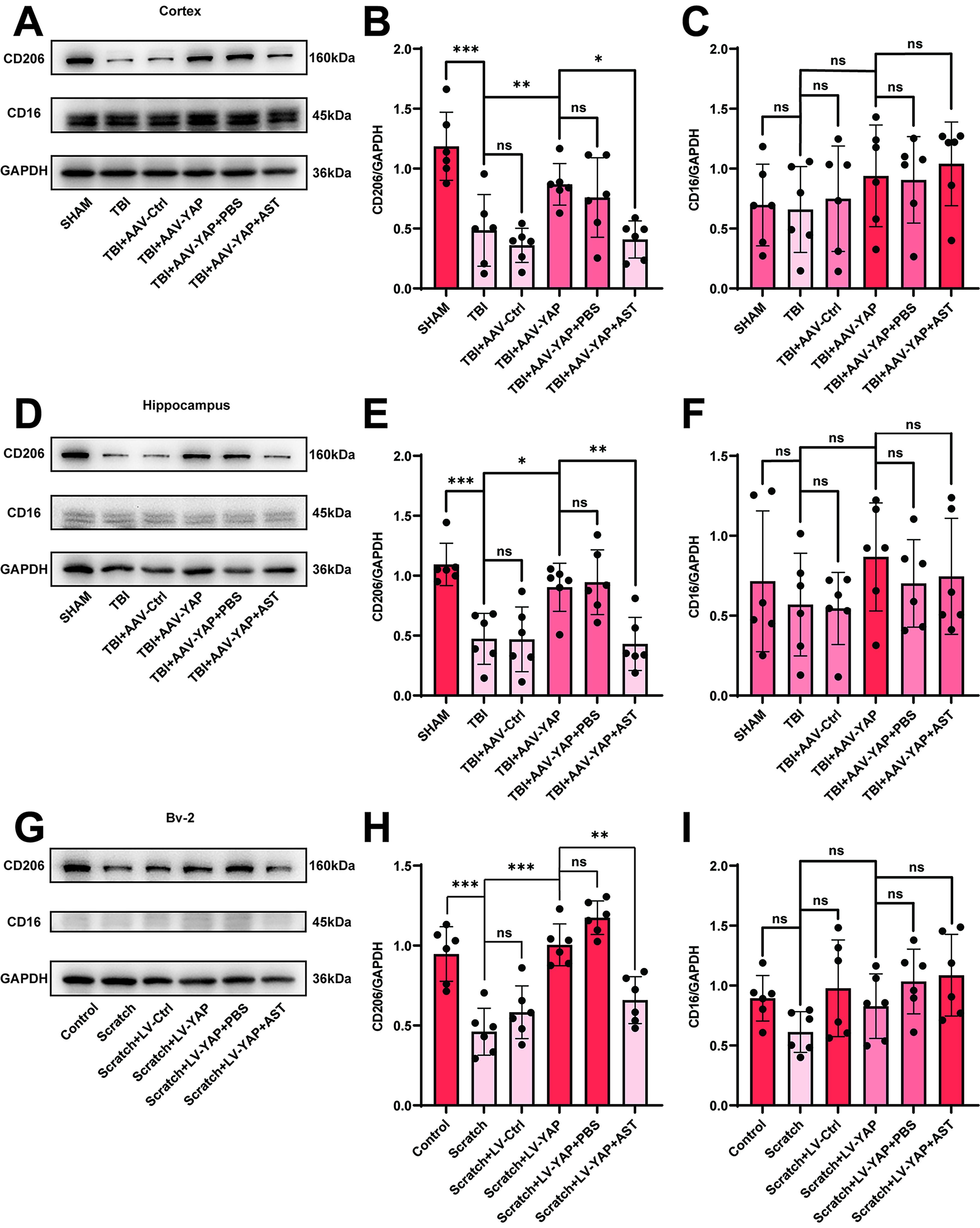
Overexpression of YAP promoted the anti-inflammatory responses of microglia
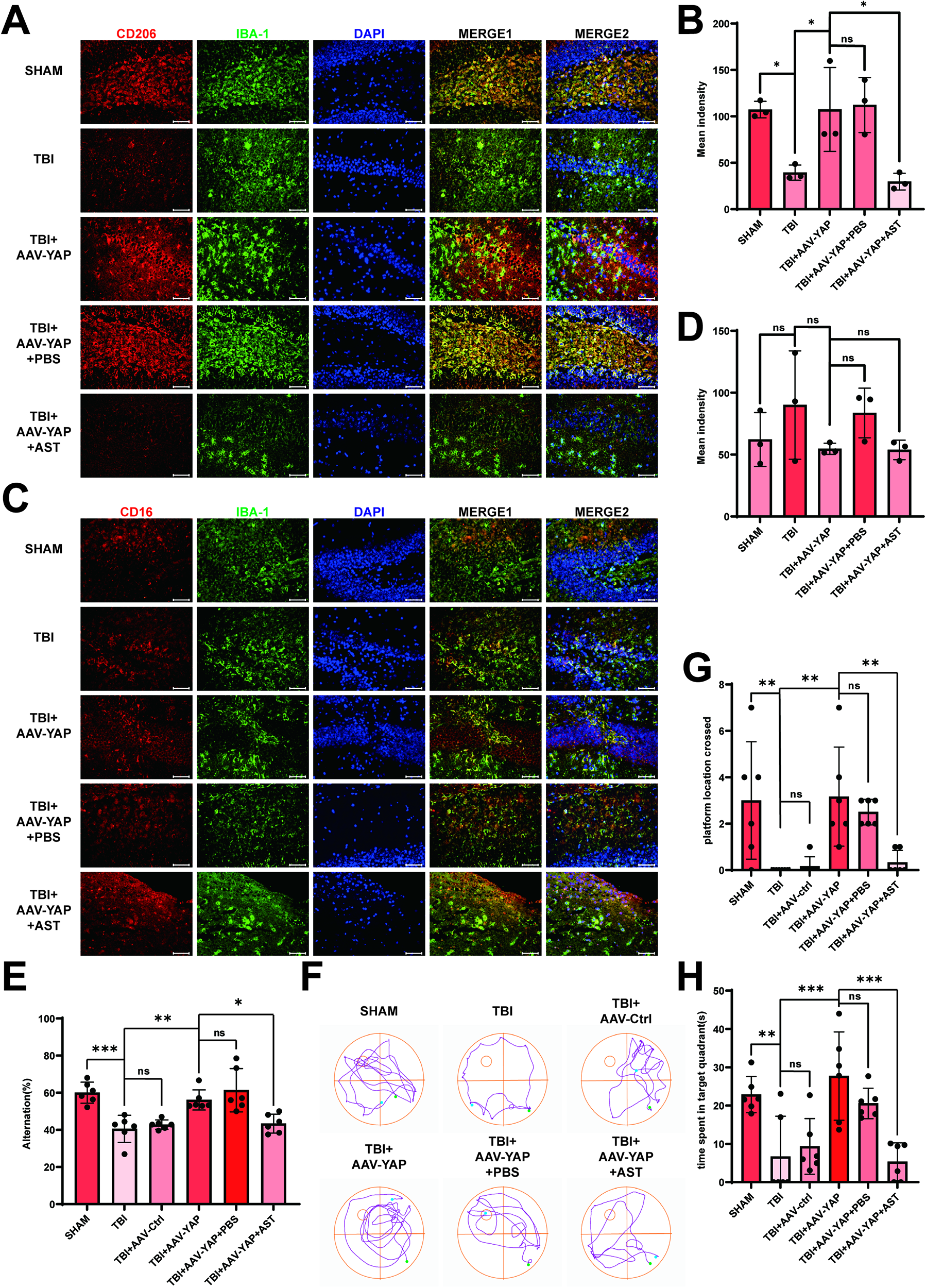
Overexpression of YAP promoted anti-inflammatory responses of microglia and improved cognitive function after TBI. (
Cognitive function was evaluated using the Y-maze and MWM tests. The TBI + AAV-YAP group showed significantly higher spontaneous alternation in the Y-maze compared with the TBI group, indicating improved spatial working memory (Fig. 8E). In the MWM test, mice overexpressing YAP demonstrated enhanced learning capability compared with the TBI group (Supplementary Fig. S7E) and spent more time in the target quadrant and crossed the platform location more frequently than the TBI controls (Fig. 8G,H), as no significant difference in swimming speed was observed among the groups (Supplementary Fig. S7F). These results suggest that YAP overexpression enhances anti-inflammatory responses in microglia and promotes cognitive recovery after TBI.
Astegolimab blocks the YAP-driven IL-33/ST2 activation
To determine whether YAP promotes anti-inflammatory responses through the IL-33/ST2 pathway, we administered astegolimab, a specific ST2 receptor blocker, 30 min before TBI induction. Treatment with astegolimab did not alter the expression levels of YAP or IL-33 in either the in vivo or in vitro models (Fig. 6E,G–K), indicating that its action is downstream of IL-33 expression.
However, astegolimab significantly attenuated the anti-inflammatory effects induced by YAP overexpression. In both cortical and hippocampal tissues, the increase in CD206 expression observed in the TBI + AAV-YAP group was reversed by astegolimab treatment (Fig. 7A–F). Similarly, in the co-culture system, the elevated CD206 expression in microglia incubated with YAP-overexpressing astrocytes was abolished by astegolimab (Fig. 7G–I). CD16 expression remained unaffected across all conditions.
Notably, the expression of the ST2 receptor itself did not differ significantly between the Sham and TBI groups (Supplementary Fig. S4E–L), suggesting that TBI does not alter ST2 availability, but that IL-33/ST2 signaling is modulated through ligand expression and binding.
Cognitive benefits associated with YAP overexpression were also counteracted by astegolimab. Mice in the TBI + AAV-YAP + AST group exhibited reduced spontaneous alternation in the Y-maze (Fig. 8E) and decreased learning capability, platform crossings, and target quadrant time in the MWM test (Supplementary Figs. S7E, 8F–H) compared with the TBI + AAV-YAP group. Meanwhile, no significant difference in the swim speed was observed in mice following AST intervention (Supplementary Fig. S7F). These findings confirm that astegolimab inhibits YAP-driven recovery by blocking IL-33/ST2 signaling.
We further examined proteins associated with the NF-κB signaling pathway downstream of the IL-33/ST2 axis in the microglial model.40,41 The level of phosphorylated NF-κB (p-NF-κB) was significantly increased in the Scratch group compared with the control group. In contrast, the co-culture system with YAP-overexpressing astrocytes (Scratch + LV-YAP group) showed a marked reduction in p-NF-κB levels relative to the Scratch group. However, when ST2 receptor signaling was blocked (Scratch + LV-YAP + AST group), p-NF-κB expression was significantly elevated again (Supplementary Fig. S8A–C).
Taken together, these results demonstrate that the impairment of cognition after TBI was considerably restored following overexpression of YAP in astrocytes, while Astegolimab reversed the effect of YAP overexpression on the activation of IL-33/ST2 signaling.
Discussion
In this study, we investigated the potential effects of modulating YAP on brain neuroinflammation and cognitive dysfunction in mice after TBI and explored the underlying mechanisms. Our results showed that the overexpression of YAP using AAV increased IL-33 expression in the injured cortex and hippocampus of mice, thereby activating the IL-33/ST2 signaling pathway in microglia and enhancing anti-inflammatory responses of microglia after TBI. However, the ST2 inhibitor astegolimab reversed the beneficial effects of AAV-YAP, worsened cognitive impairment, and reduced microglial anti-inflammatory responses without altering ST2 expression. In conclusion, YAP regulates microglia anti-inflammatory responses and neuroinflammation after TBI through the IL-33/ST2 pathway, thereby improving cognitive outcomes after TBI (Fig. 9).
TBI is a neurological disease with a poor prognosis, which is usually accompanied by severe cognitive impairment. Researchers mostly focused on secondary injury after TBI to find the best window period for treatment and intervention, and one of the pathological feature of secondary injury is neuroinflammation.1,6,42 According to previous studies,34,35 microglia play a central role in neuroinflammation, and the balance between their pro- and anti-inflammatory responses significantly influences TBI prognosis. Therefore, CD206, an indicator of anti-inflammatory responses, was used as an indicator of neuroinflammation in this study. We found that the post-TBI decrease in microglial CD206 expression was reversed by both IL-33 administration and AAV-mediated overexpression of the Yap1 gene. However, none of the other indicators showed statistically significant differences. To validate the reliability of microglial markers, we have performed quantitative co-localization analysis with Pearson’s correlation coefficient for the fluorescence images of CD16, CD206, and ST2, each co-localized with IBA-1 (Supplementary Fig. S9A–C). According to the researchers’ results, 5 the effects of IL-33 on neuroinflammation were found to be dual, which could explain the variable expression of other inflammatory markers in our in vivo and in vitro TBI models. Thus, we tried to find out upstream regulators of IL-33 secretion.
YAP is a key transcription factor in the Hippo pathway. It has been proven that the inhibition of Hippo is important for the recovery of TBI. 19 Hippo pathway activation leads to YAP phosphorylation, preventing its nuclear translocation and transcriptional activity.18,36 In our study, YAP expression was found to be decreased after TBI in both clinical brain tissue samples (Fig. 1A,B,1F,G) and TBI models (both in vivo and in vitro) (Fig. 5). Coincidentally, IL-33 also showed a similar trend (Fig. 1A,B, 1H–I and Fig. 2). Previous evidence suggests that YAP upregulation or Hippo pathway inhibition exerts anti-neuroinflammatory effects. 43 According to previous study, YAP acts as a co-transcription factor to promote IL-33 gene promoter activation in the inflammatory mode of myocardial infarction (MI). 21 We also found that the nuclear content of YAP was also decreased after TBI, but after the overexpression of YAP by lateral ventricular injection of AAV or by lentiviral transfection in astrocytes, we explored the mechanism by which YAP overexpression increased the anti-inflammatory responses of microglia after TBI. Also, we found YAP protein effectively binds to the promoter region of the IL-33 gene (Supplementary Fig. S5). As expected, the expression level of IL-33 increased after upregulating YAP (Fig. 6E,G–K), as well as the expression of anti-inflammatory indicators of microglia increased (Fig. 6E–F, Fig. 7, and Fig. 8), indicating that YAP is a key factor driving the expression of IL-33 secreted by astrocytes and stimulating anti-inflammatory responses of microglia.
Astegolimab is a specific antagonist of the IL-33 orphan receptor ST2 in clinical application, and there is no relevant literature on its use in the nervous system. To confirm that the facilitation of overexpressed astroglial YAP on microglial anti-inflammatory responses was dependent on the IL-33/ST2 pathway, we injected astegolimab into the lateral ventricle of TBI model mice after the administration of AAV-YAP. The results of Western blotting showed that the expression level of ST2 did not change (Supplementary Fig. S4E–L); at the same time, the drug reversed the anti-inflammatory responses of microglia and improvement of cognitive function. As a key indicator of NF-κB pathway activation, the altered p-NF-κB levels reflect functional changes in microglia during neuroinflammation (Supplementary Fig. S8A,B), which is consistent with the corresponding changes we observed in the anti-inflammatory marker CD206(Fig. 8G–H). However, the specific interaction between the signals transmitted by ST2 receptors and the signaling pathways controlling the anti-inflammatory responses of microglia have not been fully revealed, which may require further exploration.
Conclusions
Taken together, our data demonstrate that YAP exerts a protective effect on neuroinflammation after TBI through the IL-33/ST2 pathway. Our results provided a potential target to promote the anti-inflammatory responses of microglia as well as novel therapeutic ideas for the future treatment of secondary brain injury after TBI.
Transparency, rigor, and reproducibility summary
The study and analysis plan were registered before beginning data collection with the National Natural Science Foundation (No. 81771292) and Natural Science Foundation of Jiangsu Province (No. BK20221557). Although the analysis plan was not formally preregistered, the team member primarily responsible for the analysis, M.-L.Z., verifies that the plan was prespecified. We planned for a sample size of 280 subjects based on the availability of mice. Out of these, data from 219 mice were usable. Among the mice that generated usable data, a total of 51 mice were used for immunofluorescence analysis, 102 mice were used to assess the levels of protein, and 66 mice were used for the Y-maze test and Morris water maze. The remaining 61 mice did not yield usable data because they died during the induction phase of the TBI model, which involved anesthesia and hypothermia. In regard to the molecular biology experiments, each mouse sample underwent six technical replicates in the in vivo studies, and the average was calculated. The six average values obtained from the six mice in each group (n = 6/group) were used for statistical comparisons between groups. In the in vitro experiments, each experiment was conducted in two wells to obtain an average count, and three independent biological replicate experiments were performed. Regarding the data analysis for the pathological experiments, each group consisted of three mice, and three coronal brain slices from the injured area were taken from each mouse. Under fluorescence microscopy, we selected one field of view surrounding the injured area for quantifying the detection indicators and averaged the data to represent the experimental data for each mouse. The average values for each group (n = 3/group) were then used for statistical comparisons between groups. Investigators blinded to the participants’ relevant characteristics performed biomechanical quality control decisions and analyses. All mice were tested between 08:00 and 12:00 in a fed state. Histological analyses were conducted in three batches, with samples randomly assigned to batches. We established negative controls for positive results by setting up a solvent group and a sham operation group. All equipment and analytical reagents used for experimental manipulations and measurements may be available upon request from commercial sources (details can be found in the Materials and Methods section). To the best of our knowledge, no replication or external validation studies have been performed or are planned at this time. The data that support the findings of this study are available from the corresponding author upon reasonable request.
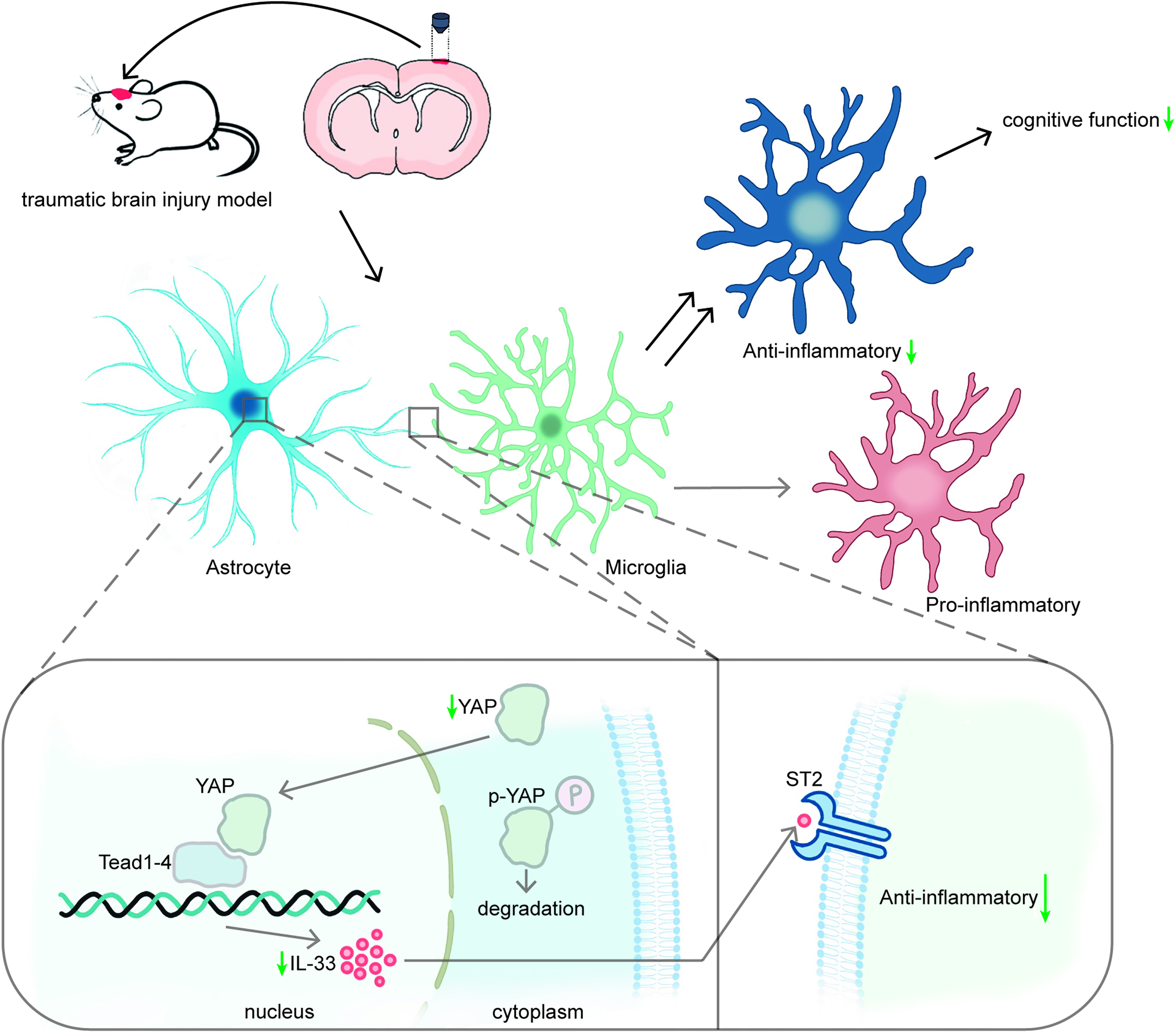
Graphical abstract showing the molecular mechanism of the changes in neuroinflammation after TBI. YAP regulated the anti-inflammatory responses of microglia through mechanisms mediated at least partially by the IL-33/ST2 pathway after TBI.
Authors’ Contributions
M.L.Z., R.Z., S.Q.G., and X.W. designed and interpreted the investigation. R.Z., X.B.Z., Y.S., and W.X.J. performed the experiments. M.L.Z., J.Y.Q., C.C.G., T.L., and Y.L.H. analyzed the data. R.Z., X.W., S.H.M., and S.Q.G. wrote the article. All authors read and approved the final article.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the National Natural Science Foundation of China (No. 81771292) and Natural Science Foundation of Jiangsu Province (No. BK20221557).
Data Availability
All data that support the findings of this study are available from the corresponding author upon reasonable request.
Ethics Approval
All procedures involving animals were performed in accordance with the Guidelines for the Care and Use of Laboratory Animals published by the National Institutes of Health and approved by the Animal Care and Use Committee of Jinling Hospital. Human brain tissue and blood were collected using a protocol approved by the Ethics Committee of Jinling Hospital (approval No. 2024DZGJJ-190).
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.