
Abstract
Traumatic brain injury (TBI) has systemic consequences for patients, including a serendipitous role in enhancing fracture healing. Although most polytraumatic injuries impair bone repair, TBI has been associated with accelerated fracture healing and excessive callus formation. This review explores the current understanding of brain–bone interaction and the mechanisms by which TBI may promote osteogenesis. Key contributing factors include an altered immune response, endocrine modulation, sympathetic signaling, neuropeptide signaling, increased osteogenic factors, and exosomal microRNAs. These components influence many elements of fracture healing, including macrophage polarization, osteoblast differentiation, angiogenesis, and suppression of osteoclast activity. Additionally, the overlap between mechanisms of neurogenic heterotopic ossification and fracture healing in the context of associated TBI will be reviewed. Despite substantial pre-clinical and clinical evidence supporting this phenomenon, its translation to therapeutic strategies remains limited. We will discuss future directions for fracture studies that consider the emerging mechanisms of TBI-induced accelerated fracture repair, the existing complexity and challenges in the field, and the potential role of the evidence in developing novel therapeutic options. Understanding these pathways holds promise for advancing fracture and complex musculoskeletal injury treatment, ultimately improving patient outcomes.
Introduction
Polytraumatic injuries, particularly those involving both the brain and the skeletal system, pose a unique challenge to clinicians and researchers due to the complex healing processes of multiple injured organs. 1 Approximately 50 million people sustain a traumatic brain injury (TBI) each year globally, and half of the world’s population experiences at least one TBI in their lifetime. 2 Given that the prevalence of significant extremity injuries in polytrauma patients is nearly 60%, 3 many patients with TBI experience a simultaneous fracture. TBI has been reported to occur in up to 30% of patients with upper extremity injuries, 17% of patients with tibial fractures, and 28% of patients with femoral fractures. 4 This underscores the importance of gaining further understanding of how the brain and bone interact in polytraumatic conditions.
Polytrauma, historically defined as significant injuries in two or more locations, 5 is clinically associated with impaired bone healing and increased rates of nonunion.6–8 Murine models provide further evidence, demonstrating that chest trauma impairs fracture healing9,10 and leads to greater polymorphonuclear neutrophil (PMN) infiltration at the fracture site. 11 Findings from our previous study suggest that an exacerbated and prolonged inflammatory phase induced by chest trauma dysregulates fracture repair. 1 Additionally, burn injury impairs fracture healing through immune response modulation. 10
In contrast to the general trend of impaired fracture healing in polytrauma, central nervous system (CNS) injury appears to enhance fracture repair. Long-term effects of TBI are the reduction of overall bone mineral density and an increased risk of fractures.12,13 However, TBI with a concomitant fracture results in accelerated bone healing and increased callus formation. 14 Spinal cord injury similarly accelerates fracture healing.15,16 Given these findings, insights into the cellular and molecular mechanisms of brain–bone cross talk during TBI polytrauma may pave the way for future breakthroughs in fracture repair.
Accelerated fracture healing and excessive callus formation in the context of TBI are well documented. The phenomenon was initially described in 1964 17 with subsequent studies providing further support.18,19 In 2005, the first review article published about the topic stated that the positive effect of TBI on fracture healing remained uncertain. 20 Ten years later, an updated review article confirmed that TBI-induced fracture healing is generally accepted by the scientific community. 21 Murine models show increased callus formation and bone density in TBI with fracture groups compared to isolated fracture groups,22,23 and clinical studies demonstrate that patients experiencing head trauma exhibit faster rates of fracture healing with higher callus volume.24–27 TBI was associated with a twofold increase in callus volume after 2 weeks and a 90% union rate at 4 weeks, compared to 60% union in controls.23,28 In vitro studies have also revealed that serum from patients and mice experiencing combined TBI with fracture has higher osteogenic cell proliferation as compared to fracture-only serum.29,30 CNS injury increases the likelihood of heterotopic ossification, defined as the abnormal formation of extraskeletal bone (heterotopic ossification) in soft tissue. 31 Although the exact relationship between heterotopic ossification and faster fracture healing in TBI remains unclear, several mechanisms have been proposed to explain why TBI accelerates bone repair. These include how TBI induces endocrine factors,26,32 sympathetic signaling, 33 neuropeptides, 34 osteogenic factors,35,36 microRNA (miRNA), 37 and immune modulation 38 resulting in an accelerated fracture healing process.
Brain tissue mediates homeostasis through cross talk with different organ systems, and bone plays a key role in mineral storage, hematopoiesis, endocrine functioning, and mechanical support. 39 Polytrauma research has shed light on the interaction between these two seemingly unrelated organs. Despite recent advancements in understanding the intricacies of brain–bone cross talk in polytrauma over the past few decades, much remains to be uncovered about how these two systems interact. Although the most recent review article surrounding the topic by Xiong et al. comprehensively reviews several mechanisms of brain injury–induced fracture healing, 40 there are still emerging areas of study such as TBI and miRNA expression and relationships to heterotopic ossification, which remain active areas of investigation. In this review, we begin by reviewing the normal fracture healing process. Thereafter, the most prominent mechanisms of bone–brain interaction in polytrauma and alterations in fracture healing are described. Based on the current mechanistic understanding, future directions and treatment modalities are then discussed.
Physiology of Bone and Fracture Healing
Bone is comprised of a mineral component that imparts stiffness and is surrounded by an organic matrix that allows for flexibility and elasticity. 41 The mineral substance primarily consists of crystalline hydroxyapatite formed from the nucleation of calcium and phosphate ions. Magnesium, bicarbonate, sodium, zinc, citrate, carbonate, barium, strontium, fluorite, and potassium also reside in the inorganic material.42,43 The organic component includes mostly type I collagen and noncollagenous proteins including fibronectin, osteocalcin (OCN), osteonectin, osteopontin, bone sialoprotein, bone morphogenetic proteins (BMPs), and growth factors. 44 Within the extracellular matrix, the main types of cells include osteoblasts, osteoclasts, and osteocytes.42,43 Osteoblasts arise from mesenchymal stem cells (MSCs) and function to form bone by secreting osteoid and matrix proteins. 44 Osteoclasts are derived from mononuclear monocyte precursor cells and function to resorb bone by secreting H+ and the enzyme cathepsin K. 45 Osteocytes, derived from osteoblast differentiation, have an array of functions including mechanosensation, supporting bone structure, and regulating mineral metabolism. 46
Although brain and bone may appear as separate entities that do not significantly affect each other, research has proven that the two organs are interconnected.47–49 Brain–bone cross talk is especially evident in fracture healing. Typically, fracture healing involves an inflammatory phase, a repair phase, and a remodeling phase. 50 The inflammatory phase is characterized by fracture, induced blood vessel rupture, and hematoma formation accompanied by PMN and macrophage infiltration. 51 In the repair phase, callus formation and collagen matrix deposition occur, and in the remodeling phase, bone is restored to its normal structure. 52 Both clinical studies25,53 and murine models23,38,54 show that TBI significantly alters the normal fracture healing process through a variety of factors that will be discussed in detail in the following sections.
Endocrine Factors
Several key endocrine factors and their roles have been identified in TBI and expedited fracture healing. One of the most prominent is leptin, which was first discovered in 1995. 55 Leptin is extensively studied for its role in obesity and satiety, 56 whereas its involvement in bone metabolism has emerged as a more recent area of research. Leptin has effects on bone metabolism peripherally to promote osteogenesis.57–59 Once released from fat, including bone marrow adipocytes, leptin can achieve its peripheral osteogenic effect by directly stimulating growth of osteoblasts and chondrocytes. 60 Centrally acting leptin was initially thought to impair osteogenesis. 61 However, central leptin gene therapy has resulted in higher levels of bone formation in leptin-deficient mice. 62 In addition, Bartell et al. 63 reported that intracerebroventricular injection of leptin increased bone formation and mineral density in leptin-deficient mice. 63 Studies have also proposed that leptin may indirectly improve fracture healing by increasing levels of growth hormone and insulin-like growth factor 1, which are potent stimulators of bone growth.64,65 Taken together, peripheral leptin has a predominantly osteogenic effect through direct stimulation, whereas central leptin may indirectly induce bone formation. 66
Given this influence of leptin on bone metabolism, studies have begun to correlate leptin’s osteogenic effects with TBI-induced accelerated fracture healing (Table 1). In murine models, TBI resulted in higher levels of cerebrospinal fluid leptin, likely as a result of the breakdown of the blood–brain barrier. 64 Additionally, some studies established that more rapid fracture healing after TBI does not occur in leptin-deficient mice,32,70 which was rescued after the administration of exogenous leptin. 64 A secondary analysis of a clinical study by Khallaf et al. also reported an early increase in leptin levels in patients with combined TBI and fracture compared to controls.4,25 These findings suggest that leptin plays an integral role in increased bone healing after TBI.
A Summary of Recent Evidence on the Impact of Traumatic Brain Injury on Fracture Healing
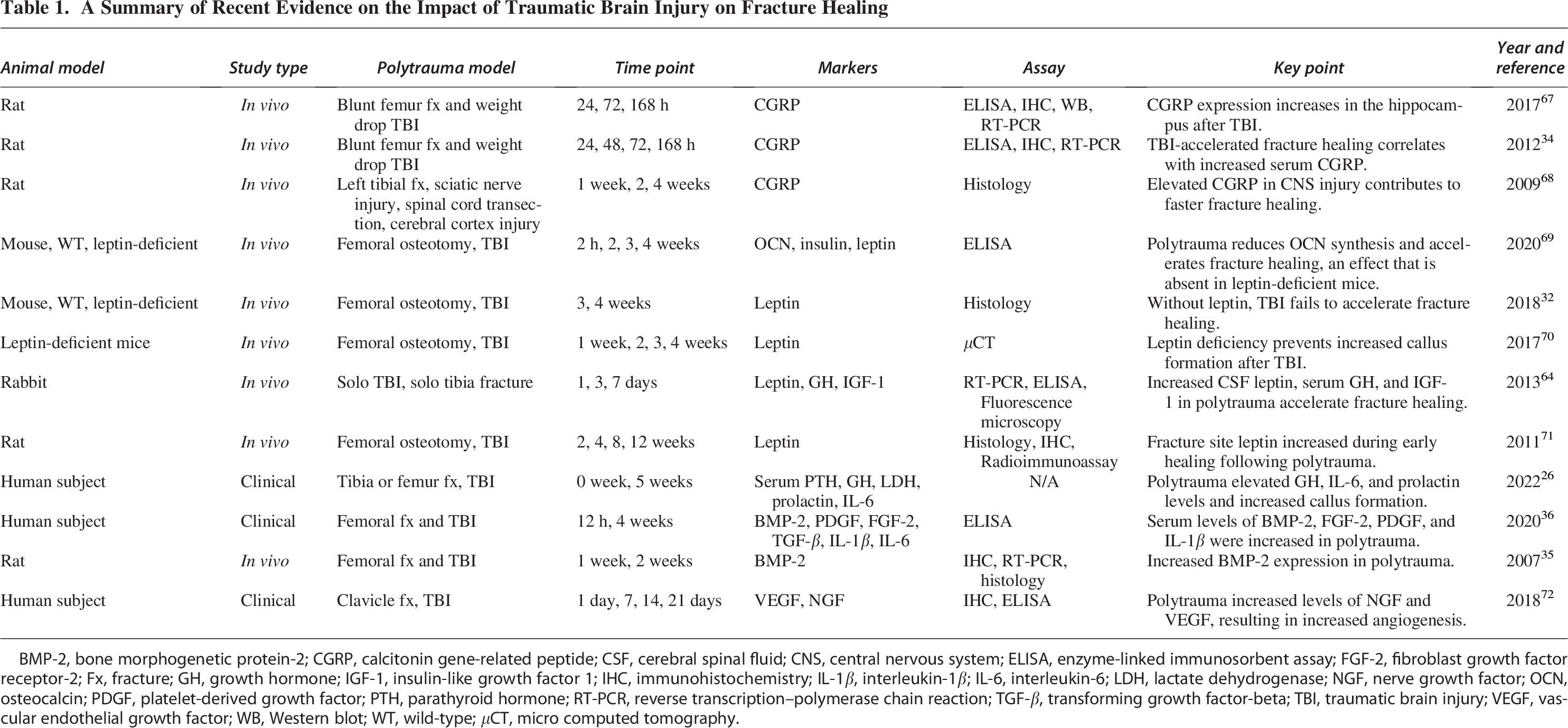
BMP-2, bone morphogenetic protein-2; CGRP, calcitonin gene-related peptide; CSF, cerebral spinal fluid; CNS, central nervous system; ELISA, enzyme-linked immunosorbent assay; FGF-2, fibroblast growth factor receptor-2; Fx, fracture; GH, growth hormone; IGF-1, insulin-like growth factor 1; IHC, immunohistochemistry; IL-1β, interleukin-1β; IL-6, interleukin-6; LDH, lactate dehydrogenase; NGF, nerve growth factor; OCN, osteocalcin; PDGF, platelet-derived growth factor; PTH, parathyroid hormone; RT-PCR, reverse transcription–polymerase chain reaction; TGF-β, transforming growth factor-beta; TBI, traumatic brain injury; VEGF, vascular endothelial growth factor; WB, Western blot; WT, wild-type; μCT, micro computed tomography.
Prolactin, an endocrine factor known for its role in lactation, has also been associated with fracture healing after TBI. 73 Ravi et al. 26 found that the mean value of serum prolactin levels in patients was higher 5 weeks after TBI and fracture polytrauma as compared to isolated fracture, suggesting prolactin may influence fracture healing. 26 Although there is emerging evidence that acute elevations in prolactin, such as those seen after traumatic injury, may contribute to osteogenesis, the underlying mechanisms remain poorly understood.
Sympathetic Signaling
TBI is frequently accompanied by a pronounced hyperadrenergic state characterized by elevated circulating catecholamine levels.74,75 This response is widely considered part of a systemic stress reaction aimed at maintaining physiological homeostasis following severe injury, as activation of the sympathetic nervous system enhances peripheral release of catecholamines such as norepinephrine (NE). 76 Notably, plasma NE concentrations rise in proportion to injury severity, demonstrating a dose-dependent relationship with the extent of neurological trauma. 77
Jahn et al. 33 found that although TBI-induced sympathetic stimulation impairs bone formation in intact bone, sympathetic signaling promotes fracture healing through angiogenesis. 33 Specifically, osteogenic type-H vessel proliferation occurred in the fracture callus of mice treated with the β2 adrenergic receptor (ADRB2) agonist formoterol. They found that when NE binds ADRB2, it stimulates the expression of the neuropeptide CGRP, resulting in increased vascular endothelial growth factor (VEGF) and ultimately vessel proliferation. 33 A retrospective cohort analysis of 72 patients with long bone fractures was conducted and supported their findings, revealing improved callus formation in those who received intravenous NE. 33
Sympathetic stimulation also influences hematopoiesis, as bone marrow is richly innervated by sympathetic nerve fibers, which allows the CNS to regulate stem and progenitor cell behavior through catecholaminergic signaling. 78 β2 adrenergic signaling within the marrow niche has been shown to bias hematopoiesis toward anti-inflammatory myeloid lineages. 79 This upstream modulation of immune cell output may partially explain the increased prevalence of M2 macrophages and accelerated transition from inflammation to repair observed during fracture healing after TBI. 79 The altered immune environment after TBI is further discussed in the immune response to fracture healing section. Taken together, these studies suggest that increased sympathetic outflow after TBI plays an essential role in accelerated fracture repair. NE is therefore a potential therapeutic option for those suffering from long bone fractures.
Neuropeptides
Although sympathetic signaling has been implicated in fracture repair, nonsympathetic sensory neuropeptides such as calcitonin gene-related peptide (CGRP) and substance P are also associated with positive effects on fracture healing in the setting of TBI. CGRP, a member of the calcitonin peptide family, can be found throughout nerve fibers in the central and peripheral nervous system (PNS). 80 CGRP is synthesized in sensory nerve fibers 81 and stored in cells or released through exocytosis. 82 This neuropeptide has multiple functions including musculoskeletal pain, especially in osteoarthritis,83,84 promotion of angiogenesis, 85 vasodilation, 86 and inflammation. 87 CGRP has both neuronal and nonneuronal targets including bone. 88
As CGRP nerve fibers are abundant within bone marrow, periosteum, synovium, and surrounding tissue, they serve as a link between the nervous system and bone tissue. 80 Studies show that CGRP shares a similar role as calcitonin by inhibiting osteoclasts and therefore decreasing serum calcium. 89 CGRP can inhibit osteoclasts by reducing the expression of receptor activator of nuclear factor kappa-beta ligand (RANKL) and upregulating OCN or osteoprotegerin (OPG) in osteoblasts. 90 Exogenous human CGRP decreases osteoblast expression of RANKL in a dose-dependent manner, and this effect is diminished with CGRP receptor antagonists. 90 CGRP can also prevent RANKL-induced activation of NF-κB in bone marrow–derived macrophages, ultimately inhibiting bone resorption and osteoclastogenesis.91,92 In addition to inhibiting osteoclasts, CGRP can also promote osteogenesis. 80 In the presence of CGRP, osteoblasts increase expression of cyclic adenosine monophosphate, activating transcription factor 4 (ATF4), and OCN. 90 ATF4 is a transcriptional factor necessary for osteoblast differentiation and the transcription of OCN, a commonly used biomarker of osteoblast maturation.93–95 CGRP also has the ability to increase the expression of bone morphogenetic protein 2 (BMP2), an osteogenic factor, 96 and induce osteogenic differentiation of bone marrow stromal cells (BMSCs) in vitro. 97 Moreover, CGRP can act as an immunomodulatory peptide by inducing anti-inflammatory M2 macrophage polarization, which may help facilitate fracture healing.98,99 Finally, CGRP’s angiogenic effect is due to increased MSC expression of VEGF and fibroblast growth factor 2 (FGF-2) 100 and direct stimulation of endothelial cell proliferation. 101 The effects of CGRP are summarized in Figure 1.
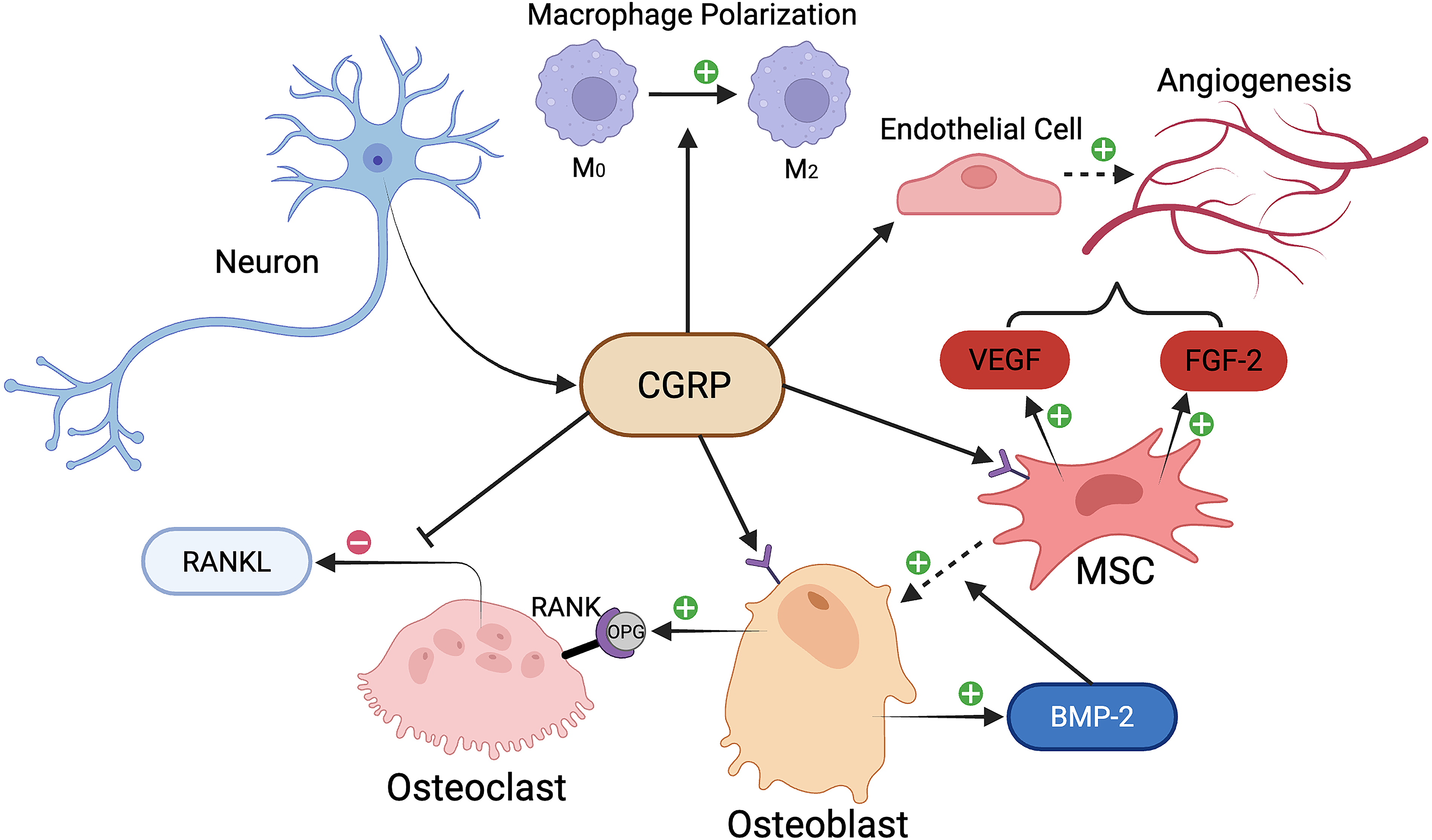
A schematic of the influence of calcitonin gene-related peptide (CGRP) on fracture healing in traumatic brain injury (TBI). The osteogenic effects of CGRP are denoted by positive and negative symbols. BMP-2, bone morphogenetic protein-2; FGF-2, fibroblast growth factor 2; OPG, osteoprotegerin; RANKL, receptor activator of nuclear factor kappa beta ligand; VEGF, vascular endothelial growth factor. Image created using BioRender.
CGRP’s ability to increase angiogenesis, inhibit bone resorption, and promote bone formation may play a large role in TBI in promoting bone repair. Song et al. 67 proposed that CGRP is expressed in the hippocampus, and as a neuroprotective agent in TBI, it acts in an autocrine or paracrine manner. 67 The expressed CGRP may enter the serum after blood–brain barrier disruption and enhance fracture healing. 67 Rats subjected to femoral fracture and TBI also have increased levels of CGRP in the choroid plexus, further suggesting that CGRP-enriched cerebrospinal fluid can leak out into the serum after blood–brain barrier disruption in TBI. 34 Additionally, CGRP was found to be elevated in the dorsal root ganglion of rats after both spinal and cerebral cortex injuries. 68 CGRP may travel from the dorsal root ganglion to the fracture site using axonal transportation. 68 This mechanism is supported by a study that reveals electrical stimulation of the dorsal root ganglion of rats accelerates osteoporotic fracture repair via increased synthesis and release of CGRP. 102 These studies are summarized in Table 1.
Similarly to CGRP, substance P is another neuropeptide that can be found in sensory nerve fibers and facilitates nociceptive signaling. 88 It acts on the neurokinin 1 (NK1) receptor and is involved in the modulation of pain, inflammation, vasodilation, and nonneuronal tissue. 88 NK1 receptors can be found on osteoblasts, osteoclasts, osteocytes, chondrocytes, and mast cells.103–105 Substance P may also be expressed by macrophages and T lymphocytes, which are heavily involved in fracture repair. 106 Along with CGRP-positive nerves in bone tissue, nerves containing substance P can be found in similar locations such as the periosteum, epiphyseal growth plate, bone marrow, ligaments, synovium, and subchondral bone. 107
The osteogenic effects of substance P arise from its ability to upregulate runt-related transcription factor 2 (RUNX2), OCN, alkaline phosphatase, and collagen type I gene expression in BMSCs via the wnt/β-catenin pathway of osteoblastic precursor cells.108–110 Substance P also increases expression of VEGF and BMP-2 in BMSCs, which both have the capability of facilitating fracture healing. 110 Although there is a lack of research connecting substance P to fracture healing in TBI, the concentration of substance P in the serum was elevated in both a rodent model 111 and a human study after TBI. 112 These findings suggest that substance P may facilitate fracture healing after CNS injury, although further studies are needed to confirm its role.
Sensory innervation and its associated neuropeptides are critical for fracture repair, which may partially explain why PNS injury, in contrast to CNS injury, tends to impair bone healing. Mice exhibit delayed callus formation and mineralization after removal of sympathetic nerve fibers and subsequent absence of substance P. 113 Furthermore, after sciatic nerve resection, there is defective callus formation in rat and rabbit animal models. 114 Inferior alveolar denervation also impairs mandibular defect healing in rats. 115 The PNS facilitates bone formation through a combination of neuropeptides, neurotransmitters, and a shift of nerve-resident stem cells toward restorative phenotypes. 114 As previously discussed, many of these benefits appear to be enhanced after CNS injury, implicating that the PNS is a key mediator of TBI-induced accelerated fracture healing.
Osteogenic Factors
Several osteogenic factors may also contribute to enhanced fracture healing after TBI. BMPs are a group of growth factors that belong to the transforming growth factor-beta (TGF-β) superfamily. Over 30 distinct BMPs have been identified, many of which are osteogenic and categorized into subgroups based on their amino acid sequences. 116 BMP-2 is a specific osteogenic BMP that is used clinically to promote fracture repair in the setting of nonunion and large bone defects. 117 BMP-2 plays a pivotal role in bone formation by regulating the expression of other BMPs and promoting the differentiation of MSCs into osteoblasts. 118 The messenger RNA (mRNA) expression of BMP-2 peaks around 24 h after fracture, and studies show it contributes to the initiation of the fracture repair cascade. 119
There is evidence that BMP-2 is upregulated after brain injury (Table 1). In a TBI–femoral fracture polytrauma rat model, the miRNA expression of BMP-2 was increased in the callus compared to isolated fracture at 1 and 2 weeks post-injury. 35 Moreover, immunohistochemistry revealed that the percentage of cells positive for BMP-2 was higher in the polytrauma groups. 35 Mollahosseini et al. 36 also found that expression of BMP-2 was significantly elevated in the serum of TBI and femoral fracture polytrauma patients at 12 h and 4 weeks post-injury. 36 Time to have bone union in patients with TBI and fracture was also significantly shorter than in those with an isolated fracture. 36 These findings indicate that increased levels of BMP-2 in TBI help facilitate expedited fracture repair.
VEGF, known for its influence on angiogenesis, 120 may also enhance fracture healing in TBI. When VEGF binds to its receptor, it leads to the recruitment of endothelial progenitor cells and the proliferation of endothelial cells, ultimately influencing the angiogenic response to fracture healing. 121 VEGF can be expressed by inflammatory cells, mesenchyme, osteoblasts, and chondrocytes in the fracture callus. 121 In a human study of clavicle fracture, increased expression of CD31, a marker for endothelial cells, and VEGF was found in the callus of patients who experienced fracture with concomitant TBI (Table 1). 72 In the same study, Zhang et al. 72 observed elevated levels of serum nerve growth factor (NGF) and increased NGF in the callus tissue of the polytrauma patients. 72 NGF is a neurotrophin that is instrumental for survival and differentiation of the PNS and integrity of cholinergic neurons of the CNS. 122 Administration of NGF in a murine femoral fracture model improves fracture healing. 123 This neurotrophin increases adhesion molecule expression and endothelial cell proliferation during the inflammatory phase of fracture healing, 124 and Zhang et al. 72 suggest that NGF may therefore induce VEGF expression. 72 Interestingly, NGF controls the expression of substance P and CGRP in the dorsal root ganglion. 125 NGF is therefore a possible modulator resulting in increased levels of these osteogenic nociceptive neuropeptides in TBI, further contributing to accelerated fracture healing.
FGF-2 and platelet-derived growth factor (PDGF) are additional osteogenic factors that are elevated in TBI and may induce fracture healing (Table 1). 36 FGF-2 can improve fracture healing through stimulation of fibroblasts. 126 Hurley et al. 127 also found that transgenic mice with overexpression of a FGF-2 isoform exhibited accelerated fracture healing, likely due to increased vascular invasion and chondrocyte and osteocyte differentiation. 127 Although some studies indicate PDGF does not directly impact bone formation, 128 PDGF is significantly increased in TBI, leading researchers to suggest it may contribute to fracture healing. 36
The described osteogenic factors contribute to bone healing through different signaling pathways. For example, once BMP binds to its receptor on osteoblasts, Smad proteins regulate the transcription of osteogenic genes through the BMP–Smad pathway. 129 Similarly, TBI also increases mitogen-activated protein kinase pathway signaling, which leads to increased osteoblast functional capacity and bone mineralization.130,131 Pre-clinical studies have also reported upregulation of the wnt/β-catenin pathway after TBI.132,133 Accumulation of β-catenin and stimulation of this pathway result in increased osteoblast activity and accelerated bone healing.134,135 As discussed earlier, substance P increases osteogenesis through activation of this pathway as well. 109 Additionally, the PI3K/Akt signaling pathway is increased after TBI due to elevated expression of specific miRNA, which is discussed in detail in the next section. 136 This pathway ultimately induces osteoblast proliferation, survival, collagen production, and mineral deposition.137,138 Xiong et al. 40 have published a review article that describes each of these pathways in further detail. 40
MicroRNA
miRNA is a subtype of RNA molecule that regulates gene expression by binding to mRNAs and preventing them from making proteins. The expression profile of miRNAs within exosomes plays a role in the enhanced fracture healing that is linked with TBI. Exosomes are defined as small (30–150 nm diameter) extracellular vesicles that can be released by cells into the surrounding environment. 139 They are formed through the inward budding of endosomal membranes and may be secreted by most cells in vitro. 139 Once secreted, they bind to their specific target cells via surface ligands. 140 Exosomes contain mRNA, miRNA, cytokines, and proteins that may be transferred from one cell to another, ultimately serving as signaling molecules that transfer genetic material and, in turn, alter cell function.141,142 The proposed mechanism of exosome transfer after TBI is outlined in Figure 2. Once the miRNA in the exosome is transferred to the recipient cell, it is then able to regulate the cell’s mRNA expression. Studies provide examples of miRNA-containing exosomes that are secreted from cells heavily involved in bone metabolism. Osteoblasts secrete exosomes that regulate osteoclast differentiation by activating the RANKL signaling pathway. 143 Additionally, exosomes released by mature osteoclasts and their precursors can regulate osteoclast differentiation. 144 An example of a specific miRNA that has osteogenic effects is miRNA-21. miRNA-21 promotes differentiation of progenitor BMSC cells into osteogenic cells via the mothers against decapentaplegic homolog 7 (SMAD7) Runx-2 pathway. 145 When added to BMSCs culture, miRNA-21 increases expression of Runx-2, OCN, osteopontin, and BMP-2. 146 Moreover, miRNA-21 knockout mice BMSCs showed weakened osteogenic potential. 145
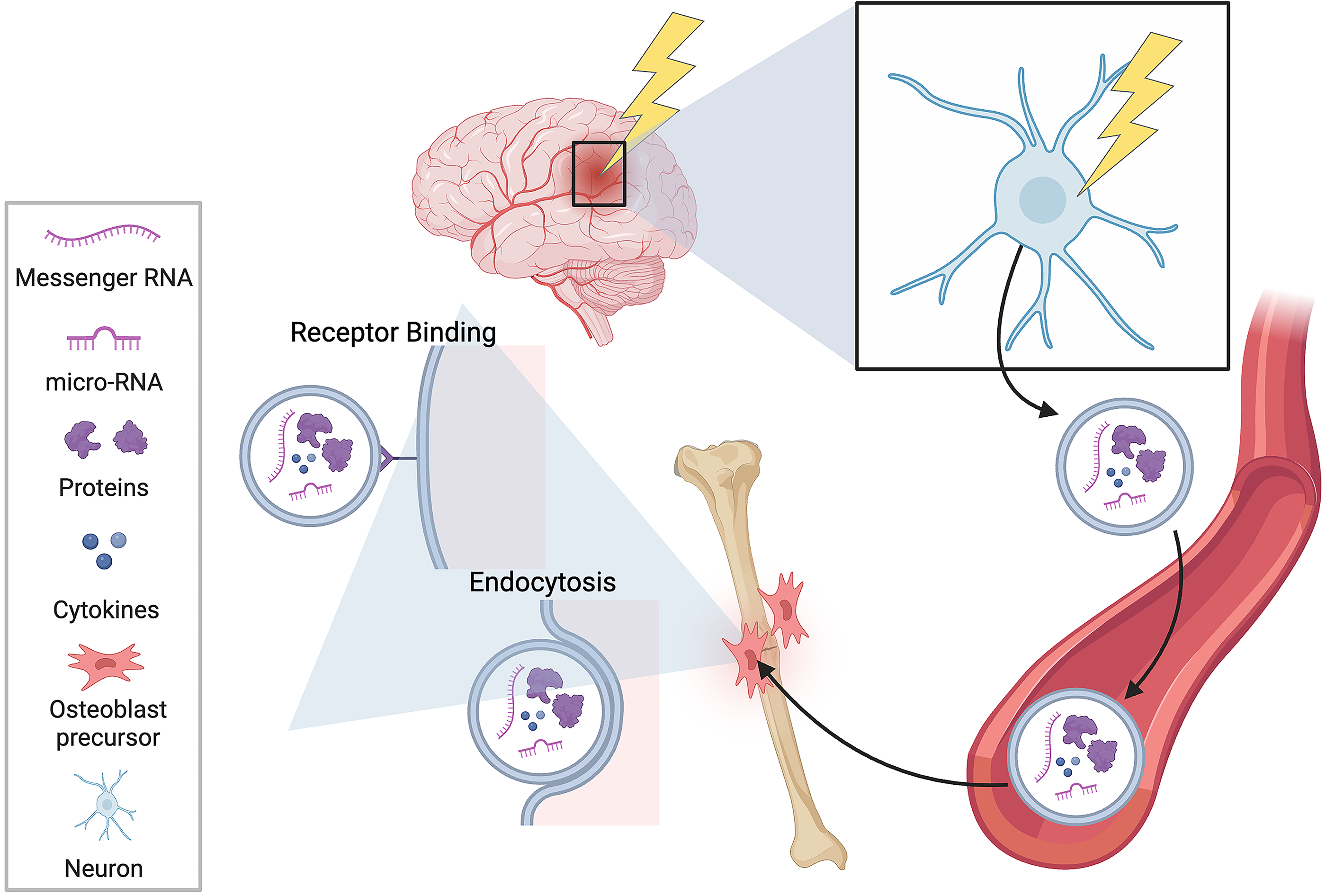
A schematic of traumatic brain injury–induced release of small extracellular vesicles. Once released into the bloodstream, vesicles are capable of binding receptors on mesenchymal stem cells and transferring genetic information through endocytosis. Image created using BioRender.
Researchers have begun to study how these miRNA-containing exosomes may change the fracture healing environment in TBI (Table 2). miRNA-21-5p, well known for its osteogenic effects, 146 was significantly increased in the exosomes and serum of patients with TBI. 37 Lin et al. 37 found that miRNA-21-5p-enriched TBI exosomes led to osteogenic differentiation in vitro via suppression of SMAD7. They also injected enriched TBI exosomes into mice after fracture and observed increased callus formation using X-ray and micro computed tomography (CT). 37 Osteogenic miRNAs such as miRNA-328a-3p and miRNA-150-5p are found in exosomes released from neurons after TBI in both rats and humans. 148 Another osteogenic miRNA, miRNA-26a-5p, is associated with the induction of osteoblast differentiation in TBI through the inhibition of phosphatase and tensin homolog, which results in increased PI3K/Akt signaling. 136 Although the concentration of the previous miRNAs described is increased after TBI, studies show that reduced levels of miRNA-433 147 and miRNA-16-5p 151 may also contribute to accelerated fracture repair.
A Summary of Recent Evidence on Traumatic Brain Injury and MicroRNA Expression
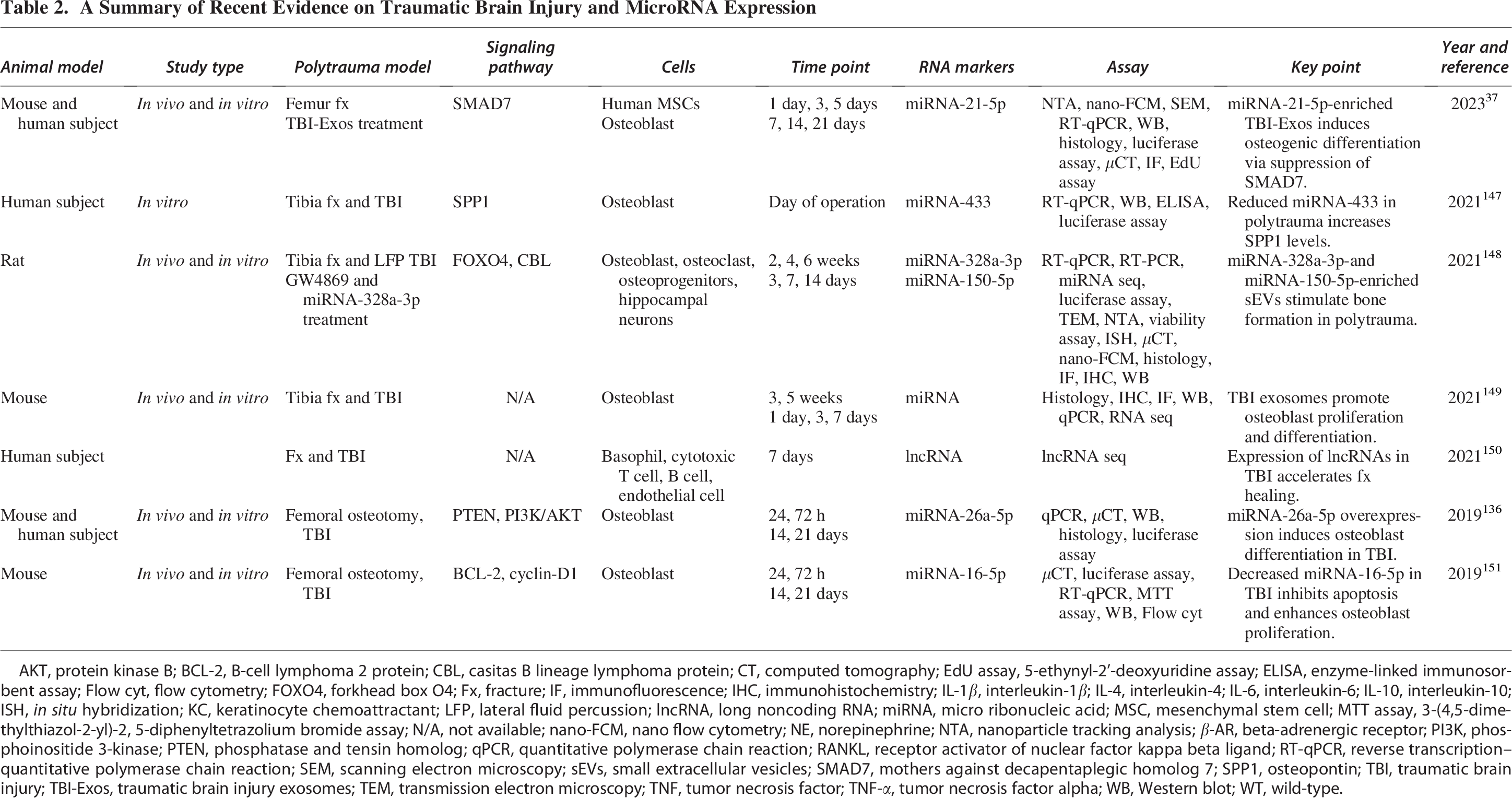
AKT, protein kinase B; BCL-2, B-cell lymphoma 2 protein; CBL, casitas B lineage lymphoma protein; CT, computed tomography; EdU assay, 5-ethynyl-2’-deoxyuridine assay; ELISA, enzyme-linked immunosorbent assay; Flow cyt, flow cytometry; FOXO4, forkhead box O4; Fx, fracture; IF, immunofluorescence; IHC, immunohistochemistry; IL-1β, interleukin-1β; IL-4, interleukin-4; IL-6, interleukin-6; IL-10, interleukin-10; ISH, in situ hybridization; KC, keratinocyte chemoattractant; LFP, lateral fluid percussion; lncRNA, long noncoding RNA; miRNA, micro ribonucleic acid; MSC, mesenchymal stem cell; MTT assay, 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide assay; N/A, not available; nano-FCM, nano flow cytometry; NE, norepinephrine; NTA, nanoparticle tracking analysis; β-AR, beta-adrenergic receptor; PI3K, phosphoinositide 3-kinase; PTEN, phosphatase and tensin homolog; qPCR, quantitative polymerase chain reaction; RANKL, receptor activator of nuclear factor kappa beta ligand; RT-qPCR, reverse transcription–quantitative polymerase chain reaction; SEM, scanning electron microscopy; sEVs, small extracellular vesicles; SMAD7, mothers against decapentaplegic homolog 7; SPP1, osteopontin; TBI, traumatic brain injury; TBI-Exos, traumatic brain injury exosomes; TEM, transmission electron microscopy; TNF, tumor necrosis factor; TNF-α, tumor necrosis factor alpha; WB, Western blot; WT, wild-type.
Immune Response to Fracture Healing
The altered immune response during the different stages of fracture healing could contribute to more rapid fracture healing after TBI. In normal fracture healing, immediately after the onset of injury, blood vessels are ruptured from the trauma. The resulting hematoma acts as a framework for the recruitment of inflammatory immune cells that secrete cytokines such as IL1, IL6, and tumor necrosis factor alpha (TNF-α). 152 Initially, the hematoma contains high levels of calcium and lactic acid, resulting in an acidic, hypoxic, and low-temperature environment. 152 The first immune cells to arrive are PMNs followed by monocytes/macrophages. 51 Mast cells also play a key role in early inflammation by recruiting neutrophils153,154 and inducing osteoclastogenesis via the inflammatory cytokine CXCL10. 155
Resolution of acute inflammation occurs as levels of inflammatory mediators decrease and immune cells are slowly cleared from the area. 156 After the initial inflammatory response, a transition to adaptive immunity follows as lymphocytes infiltrate the fracture site. 52 Pro-inflammatory M1 macrophages induce expression of RANKL from T cells and osteoblasts to activate osteoclastogenesis in preparation for callus formation/remodeling. 157 Regulatory B cells then reduce expression of pro-inflammatory mediators IFN-γ, TNF-α, and IL-2. 158 Later, a surge of the anti-inflammatory cytokines IL-4, IL-10, and IL-13 causes macrophages to polarize from the M1 inflammatory phenotype to the M2 anti-inflammatory phenotype.52,159 M2 macrophages then facilitate further suppression of inflammation and secrete IL-10, BMP-2, VEGF, and TGF-beta to aid in tissue repair, osteoblast differentiation, and angiogenesis. 160 Macrophages and neutrophils also continue to clear dead cells and debris 161 while releasing factors that recruit mesenchymal progenitor cells.162,163 This acute inflammatory phase peaks in 1–2 days and is diminished within 1 week. 52 Importantly, a special type of macrophage called osteomacs resides in bone tissue and plays a role in recognizing the original disruption and assisting bone healing during this process.164,165
At the end of the inflammatory phase, the repair phase begins, and the callus starts to form. Callus formation can occur by either endochondral or intramembranous ossification. 166 In intramembranous ossification, woven bone is formed directly from periosteal MSCs that differentiate into osteoblasts. Conversely, endochondral ossification is characterized by the initial formation of a cartilaginous soft callus template by bone marrow MSCs differentiated into chondrocytes. The mechanical environment of the fracture site determines the healing pathway and can cause recruited mesenchymal progenitor cells to undergo chondrogenic differentiation. 160 Granulation tissue can then be replaced with fibrous and cartilaginous callus, allowing for endochondral ossification. 167 Chondrocytes eventually undergo apoptosis, allowing vascular infiltration and osteoblast migration, ultimately forming a hard callus. 166 During the repair phase, there are changes in cytokine expression. Pro-inflammatory cytokines IL-1 and IL-6 are absent, 168 and TNF-α levels decrease initially and rise by the end of the repair stage. 169 The increase in TNF-α helps to initiate chondrocyte apoptosis and vascularization in endochondral ossification.169,170 After the repair phase is complete, the remodeling phase begins. Osteoblast and osteoclast activity help to restore the bone’s original structure through continuous resorption and deposition of lamellar bone.166,168 Notably, pro-inflammatory cytokines, such as IL-1 and IL-6 and TNF-α, return at this stage of healing.50,168,171
The normal immune response to a fracture is significantly altered in TBI, and this alteration likely contributes to the accelerated fracture healing seen clinically and in murine models (Table 3). Macrophage polarization is one of the key differences in TBI. Patients with clavicle fractures and concomitant TBI demonstrated a higher prevalence of M2 macrophages, faster fracture healing, and enhanced callus formation. 172 Weijian Liu et al. 79 show that increased sympathetic tone in TBI induces M2 polarization at the fracture callus of mice as a result of β2 adrenergic signaling. 79 TBI-induced M2 polarization can facilitate a more rapid transition into the anti-inflammatory repair phase and in turn enhance fracture healing. This is further supported by a study that confirms administration of IL-4 and IL-13 to induce M2 macrophage polarization accelerates fracture repair in mice. 159 Notably, the anti-inflammatory effect in TBI contrasts with other polytrauma injuries, such as chest trauma, which can induce a prolonged inflammatory response that impairs fracture healing. 1
A Summary of Recent Evidence on Traumatic Brain Injury and the Immune Response During Fracture Healing

CBC, complete blood count; CT, computed tomography; CXCL10, C-X-C motif chemokine 10; ELISA, enzyme-linked immunosorbent assay; Flow cyt, flow cytometry; Fx, fracture; G-CSF, granulocyte-colony stimulating factor; IF, immunofluorescence; IHC, immunohistochemistry; IL-1β, interleukin-1β; IL-4, interleukin-4; IL-6, interleukin-6; IL-10, interleukin-10; KC, keratinocyte chemoattractant; LFP, lateral fluid percussion; MCP-1, monocyte chemoattractant protein-1; NE, norepinephrine; 6-OHDA, 6-hydoxydopamine; OPG, osteoprotegerin; β-AR, beta-adrenergic receptor; qPCR, quantitative polymerase chain reaction; RANKL, receptor activator of nuclear factor kappa beta ligand; RT-qPCR, reverse transcription–quantitative polymerase chain reaction; sTNF-R1, soluble tumor necrosis factor receptor 1; TBI, traumatic brain injury; TNF, tumor necrosis factor; TNF-α, tumor necrosis factor alpha; UCP1, uncoupling protein 1 gene; WT, wild-type.
Additionally, there are studies that show alterations in osteoclast activity after TBI. In a murine model, osteoclastogenesis was decreased in concomitant TBI and fracture polytrauma as a result of decreased mast cells. 38 RANKL/OPG ratios are also decreased in patients experiencing a fracture with TBI, indicating an overall inhibition of osteoclasts. 173 Lower levels of osteoclast activity can lead to increased callus formation and accelerated fracture healing. Serum cytokines IL-10 and IL-6 are also increased in TBI–fracture polytrauma, indicating that cytokine expression is significantly altered.173–176 Interestingly, mice with a fracture induced contralateral to a TBI had increased callus volume compared to isolated fracture mice, whereas those with an ipsilateral TBI had similar callus volume to isolated fracture controls. 54 The study suggests that neuronal mechanisms may be more peripherally and anatomically linked to the fracture location and healing. 54 A comparison of normal fracture healing and fracture healing after TBI is outlined in Figure 3.
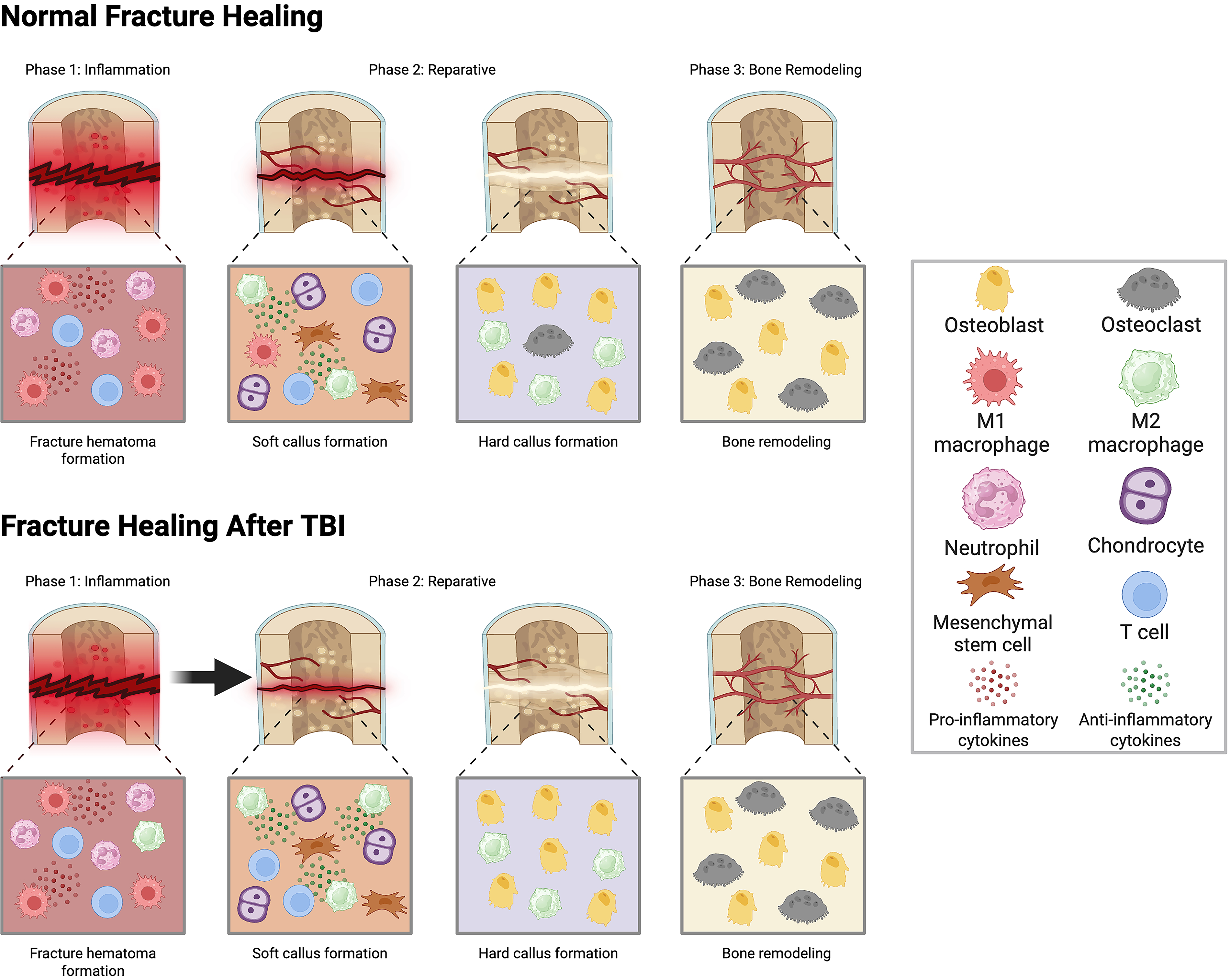
A schematic illustrating the immunological differences between normal fracture healing and fracture healing after traumatic brain injury (TBI). TBI causes a faster transition into the reparative phase due to increased M2 macrophage polarization and increased callus formation through suppression of osteoclast activity. Image created using BioRender.
TBI and Heterotopic Ossification
Heterotopic ossification is the abnormal formation of ectopic bone in soft tissues where bone does not typically exist, such as muscles, tendons, ligaments, or other connective tissues. Preceding events typically include a burn, fracture, or neurological damage. 177 Depending on the cause of heterotopic ossification, it can be classified as myositis ossificans, traumatic heterotopic ossification, or neurological heterotopic ossification. 178 TBI was first associated with heterotopic ossification in patients in 1968, uncovering the relationship between abnormal bone growth and the nervous system. 179 Since then, studies confirmed that neurogenic heterotopic ossification occurs in about 20% of patients with TBI and up to 50% of the time in those experiencing TBI–femur fracture polytrauma. 178
The mechanism of heterotopic ossification is not fully understood. One outlined mechanism is via endochondral ossification. In trauma, skeletal osteoprogenitor cells (OPCs) can combine with a microenvironment of inflammatory cells and factors, BMP signaling, and hypoxia potentially leading to endochondral bone formation.180,181 Immediately after musculoskeletal trauma with or without fracture, an influx of macrophages, neutrophils, and mast cells infiltrates the area,180,182 causing fibroblast growth factors to induce fibroblast differentiation from OPCs, forming fibrous tissue.183–185 Hypoxia-inducible factor-1 alpha and VEGF are increased as a result of hypoxia, leading to angiogenesis 186 and differentiation of chondrocytes via the transcription factor SOX-9. 187 A cartilaginous matrix template forms,188–190 then later subsides as woven bone, and eventually lamellar bone is created. 191 This process appears to share similarities with typical endochondral ossification seen in fracture healing.
CNS injury can increase the rate of heterotopic ossification through the release of osteogenic and inflammatory factors. 191 Increased blood–brain barrier permeability in TBI may increase serum levels of CGRP, BMP, FGF, IL-6, and substance P, which facilitate heterotopic bone formation through differentiation of OPCs and angiogenesis.106,191 Jiang et al. 192 found that leptin also contributes to the acceleration of heterotopic ossification through activation of the mTOR signaling pathway. 192 Although the exact relationship between heterotopic ossification and rapid fracture healing in TBI remains unclear, all the aforementioned factors involved in heterotopic ossification have been previously implicated in studies focusing on fracture healing in TBI. This indicates that the two processes are closely intertwined and provides a clear incentive for further exploration into this association.
Influence of TBI Severity on Fracture Healing
Severity of TBI may play a significant role in the fracture healing process; however, current literature on this topic is inconclusive. One study observed increased callus formation in patients with more severe head injury (GCS <7); however, interpretation of this finding is limited by potential confounders such as prolonged mechanical ventilation and duration of systemic acidosis. 26 Shim et al. 27 used the Marshall classification to assess TBI with noncontrast CT scans, reporting a strong positive correlation with severity and speed of fracture callus formation. 27 In contrast, Cadosch et al. 193 reported a negative relationship between injury severity and time to union. 193 Additionally, in a murine model, larger brain lesions with greater impact severity were associated with decreased bone formation, suggesting milder forms of TBI may preferentially accelerate fracture healing. 54 Another study found that there was no significant difference in the rate of femoral shaft fracture healing when comparing the severity of TBI or the type of intracranial hemorrhage. 53 Frequency of TBI is also an important consideration, especially for athletes at risk for repeated concussions. Although there are limited studies on this topic, repeated mild TBI impairs fracture healing in mice. 194 Given the lack of conclusive evidence, further studies that help determine how neurotrauma severity and frequency impact the fracture healing environment are needed.
Future Directions
Despite the growing body of evidence demonstrating that TBI can accelerate fracture healing, significant questions remain regarding the underlying mechanisms and how these insights might be translated into therapeutic strategies. Although this article has summarized the current proposed mechanisms of immune modulation, osteogenic factor release, neuropeptide signaling, and exosomal miRNA delivery (Fig. 4), the precise contributions and interplay between these factors remain incompletely understood.
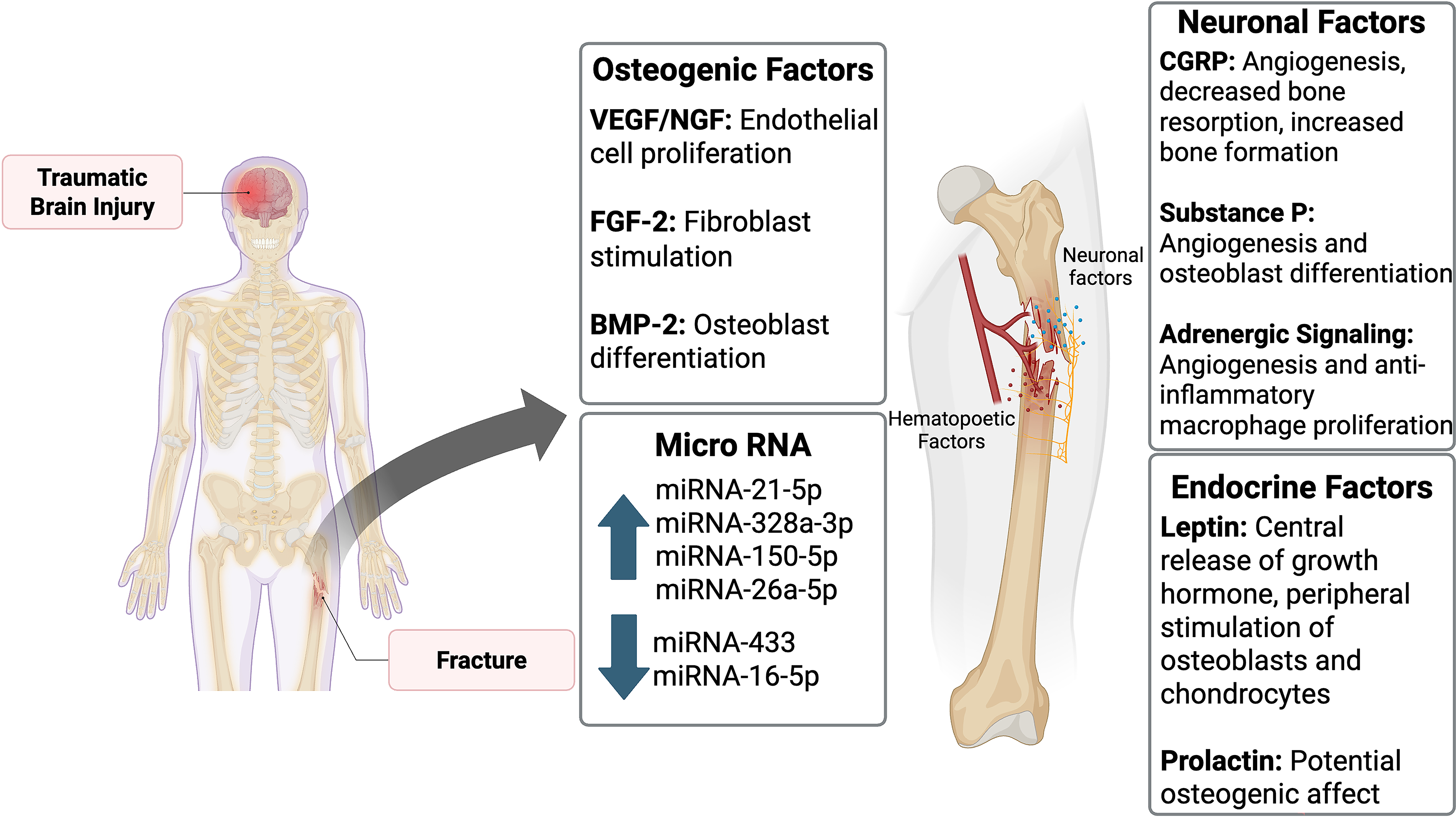
A summary of the osteogenic factors, neuronal factors, miRNA, and endocrine factors that influence fracture healing in the setting of traumatic brain injury. BMP-2, bone morphogenetic protein-2; CGRP, calcitonin gene-related peptide; FGF-2, fibroblast growth factor 2; miRNA, microRNA; NGF, nerve growth factor; VEGF, vascular endothelial growth factor. Image created using BioRender.
For example, the osteogenic role of prolactin in the acute injury setting has not been fully elucidated. Although studies discussed earlier have suggested elevation of prolactin after TBI may influence fracture healing,26,73 there is little evidence of its direct mechanistic effect. Osteoblast-like cells do express prolactin receptors, allowing for direct hormonal influence. 195 However, prolactin can cause bone turnover by increasing RANKL/OPG ratio, 196 and chronic hyperprolactinemia induces osteoporosis. 197 Further studies are needed to reveal whether the acute release of prolactin in TBI plays a significant role in stimulating fracture healing.
PDGF is another factor that has inconclusive evidence. There are studies that demonstrate that PDGF can speed up bone repair. For example, biodegradable poly(
The interplay between heterotopic ossification and accelerated fracture repair also remains speculative, suggesting a need for mechanistic studies that distinguish the two. Many of the factors released after TBI both induce heterotopic ossification and accelerate fracture healing suggesting the two are closely related. Studies aimed toward finding out the extent of the relation are needed, as the enhanced fracture healing observed may be a form of heterotopic ossification. It would be beneficial to elaborate on the possibility of forming heterotopic ossification by doing long-term follow-ups on TBI–fracture polytrauma animal models and patients. Although accelerated healing is overwhelmingly supported in pre-clinical studies, faster healing may not translate to better healing. Whether TBI improves fracture healing in the long run is still unknown, and how tunable this neuro-osteogenic response can be remains critical to prevent complications from uncontrolled ossification, heterotopic or not. Future studies analyzing late stages of bone remodeling should assess the overall biomechanical stiffness of the healed bone after TBI. Additionally, comparing different severities of TBI and subsequent fracture healing responses could be an interesting area of future research. Accelerated fracture healing persists across different pre-clinical models of TBI including cortical control impact, closed-head weight drop, weight drop with craniotomy, and lateral fluid percussion injury, yet studies comparing the severity of injury are lacking. 199
Although pre-clinical literature supports the notion that TBI accelerates fracture healing, this phenomenon is yet to be verified in large clinical studies. A decade ago, Hofman et al. 21 suggested that a prospective study with a large population size should be incentivized. 21 Although there have been several clinical articles published since then, there has yet to be a study published like what Hofman et al. 21 recommended.
Aside from CNS injury, there are no other forms of injury that show accelerated fracture healing. Our previous study revealed that chest trauma dysregulates the immune response in a way that significantly impairs fracture healing, characterized by increased innate immune cell infiltration and systemic hyperinflammation. 1 Although chest injury prolongs the inflammatory phase of fracture healing, TBI reduces inflammation and facilitates transition into the repair phase. Further characterization of immune cell phenotypes at the fracture site after TBI may help uncover the cellular mechanism behind this difference.
Improvements in our overall understanding of why CNS injury increases the speed of fracture healing may pave the way for the future of polytrauma treatment. As discussed, induction of M2 macrophage polarization via IL-4 and IL-13 enhances fracture repair in pre-clinical models. 159 Cytokine delivery systems can mimic the suppression of inflammation induced after TBI and may help improve fracture healing in patients with severe polytraumatic injury or systemic hyperinflammation. Therefore, patients with chest trauma may benefit from cytokine delivery and local immune suppression at the fracture site. Increased β2 adrenergic signaling, which can be facilitated by NE, has a similar effect on macrophage polarization and can serve as an alternate therapeutic option to modulate the immune system. 79
Another therapy that has worked in pre-clinical models is treatment with TBI miRNA-enriched exosomes, which stimulated osteogenic differentiation and enhanced fracture healing. 37 Exosome-based therapies could be tailored to deliver osteo-inductive signals without systemic side effects. The development of targeted nanoparticle delivery systems that deliver vesicles to MSCs may further optimize this strategy. Therapies that block early osteoclast activation, such as CGRP receptor agonists or RANKL/OPG modulation, may also mimic the effects seen in TBI polytrauma models. Furthermore, hormonal therapies targeting leptin or prolactin pathways may be explored, although these will require careful balancing due to the complex systemic effects of these hormones.
Conclusions
Although TBI has historically been associated with significant morbidity, its paradoxical enhancement of fracture healing offers an intriguing insight into bone regeneration biology. Advances in understanding brain–bone cross talk, particularly involving immune modulation, neuropeptides, exosomal miRNAs, and osteogenic hormones, have revealed several promising therapeutic targets. However, understanding and translating these insights into safe and effective clinical treatments remains a challenge. Ultimately, leveraging and controlling the regenerative effects observed in TBI may transform the treatment landscape for patients with impaired fracture healing.
Transparency, Rigor, and Reproducibility Statement
This review was conducted following a systematic approach to literature identification and synthesis. A comprehensive search was performed across PubMed/MEDLINE, Embase, Web of Science, and Google Scholar using MeSH terms and keywords related to TBI, bone fractures, and polytrauma. Two independent reviewers screened articles according to pre-determined inclusion/exclusion criteria, with discrepancies resolved by consensus or third-party adjudication. Data were extracted using standardized forms, and study quality was assessed using the Newcastle–Ottawa Scale for observational studies. Given substantial heterogeneity in study designs, populations, and outcome measures, a narrative synthesis approach was employed rather than meta-analysis. All data are derived from published sources cited within the article, and the complete extraction dataset is available from the corresponding author upon reasonable request.
Authors’ Contributions
T.D.B.: Literature search, first draft writing, and revision. M.R., K.S., R.P., P.S., and S.K.: Supervision and revisions. A.M.S.: Supervision, revisions, and funding resources. All authors have read and confirmed the content of this review article.
Footnotes
Author Disclosure Statement
A.M.S. serves on committees for AO, Orthopaedic Trauma Association (OTA), AAOS, and ORS. M.R. serves on committees for ISFR ORS. P.S. serves on committees for the OTA, COA, CORS, and Osteoporosis Canada. The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this review article.
Funding Information
This project was supported with funding through AO Trauma North America (Young Investigator Research Award, Early Career Development Award) and Orthopaedic Trauma Association (Young Investigator Grant #4122) awarded to Dr. A.M.S. Dr. A.M.S. gratefully acknowledges financial support from the UC Davis School of Medicine and Department of Orthopaedic Surgery. Research reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under Award Number K08AR084594 (A.M.S.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The funders had no role in the decision to publish or preparation of the article.