
Abstract
Traumatic brain injury (TBI) increases the risk for infection, and urinary tract infection (UTI) is commonly reported in patient populations that require additional medical intervention. UTIs are one of the most prevalent bacterial infections worldwide, reflecting a significant public health threat that disproportionately affects females. UTIs are most often caused by uropathogenic Escherichia coli (UPEC) and can cause delirium-like symptoms in vulnerable patients, but the specific neuroimmune and cognitive effects of post-injury UTI remain underexplored. To address this gap in knowledge, we present a unique combination of pre-clinical models of TBI and UTI to define the biological effects of bladder infection after brain injury. We hypothesize that UTI after TBI worsens the long-term outcome of brain injury. Female mice received either a sham injury or lateral fluid percussion TBI, and 3 days post-injury (DPI), mice were inoculated transurethrally with vehicle or UPEC to recapitulate a post-injury infection. Y-maze and open-field behavioral tests were administered 6 DPI and 8 DPI, respectively. Brains and blood were collected for flow cytometry and enzyme-linked immunosorbent assay from a subset of mice 6 DPI. Brains and urinary bladders were collected for immunohistochemistry from the remaining mice 10 DPI. TBI mice displayed spatial memory deficits, which were exacerbated following UTI; however, no differences in exploratory behavior were observed after TBI or UTI. TBI significantly elevated plasma interleukin-6, but strikingly, UTI following TBI dampened this response 6 DPI. Flow cytometry revealed no significant differences in circulating immune cells 6 DPI but confirmed that TBI elevated microglial reactivity and monocyte infiltration to the brain, independent of infection. TBI and UTI differentially modulated Glial Fibrillary Acidic Protein (GFAP) and Ionized Calcium-binding Adapter Molecule 1 (Iba1) expression in the hippocampus and cortex, reflecting additive and interaction-based effects. UTI after TBI elevated microglia CD68 expression in a brain region-specific manner further demonstrating that post-injury infection alters the neuroimmune landscape. Together, these data offer novel insight into the effects of UTI after TBI, showing that UTI after TBI is detrimental to overall outcome and warrant further investigation into the signaling mechanisms between bladder infection and neuroinflammation.
Introduction
Traumatic brain injury (TBI) is a substantial health concern worldwide.1,2 Improved medical care and treatment mean that patients are surviving long after injury; however, accumulating data emphasize that outcome after TBI is modulated by previous, coincident, and subsequent immune insult (see reviews3,4). Therefore, effectively managing post-injury recovery has never been more critical. Urinary tract infections (UTIs) are common, with more than 400 million infections globally per year. 5 Notably, females are 30× more likely to get a UTI compared to males during middle age. 6 About half of all females will experience at least one UTI in their lifetime with about a quarter of those females experiencing a recurrent UTI in the months following an initial infection. 7 Since female survivors are historically underrepresented in clinical TBI studies, 8 there is an urgent need to understand how sex-biased comorbidities, including UTI, impact TBI pathology and recovery. Here, we address this need by establishing a new murine model of comorbid TBI and UTI to characterize known markers of inflammatory pathology in the brain and bladder.
Between 75% and 85% of UTIs are caused by uropathogenic Escherichia coli (UPEC), which induce ascending infection by migrating through the urethra into the bladder and binding to and invading the superficial facet cells that line the lumen (as reviewed9–11). Most bladder infections are uncomplicated and arise spontaneously. Yet patient-specific risk factors, such as a urinary catheter or stent, anatomical abnormality in the urinary tract, pregnancy, or male sex, increase the risk of complicated infection and unfavorable outcome. 12 Incidence of UTI is strongly sex biased; however, the rates of infection change over time in relation to hormonal fluctuation in both males and females. 13 Many uncomplicated UTIs resolve on their own, but outpatient medical treatment can relieve urinary and pain symptoms and prevent the development of kidney infection or pyelonephritis.14–16
Hospital-acquired infections are prevalent, and moderate-to-severe TBI patients have an increased risk and susceptibility to infection soon after injury.17,18 Clinical studies show that up to 15% of adult TBI patients experience UTI within the first year after injury, which can increase length of hospital stay and mortality due to sepsis.19,20 Concerningly, the risk of infection remains high in long-term rehabilitation and home settings. In fact, clinical studies show that 33% of adult TBI patients are readmitted to the hospital within a year after injury, and 17–20% of these readmissions are due to UTIs.17,21 Typically, older age is positively associated with readmission after TBI.22,23 There is a higher prevalence of complicated UTI in aging males, 24 which inflates readmission rates in males compared to females.18,25–27 Consequently, uncomplicated UTI in females, which is typically treated in outpatient care, may be overlooked as a critical determinant of post-TBI recovery.
Even though UTIs are a frequent clinical issue after TBI,19,20,24–27 the influence of infection on long-term recovery remains unclear. TBI causes blood–brain barrier (BBB) disruption, microglia reactivity, astrogliosis, infiltration of peripheral immune cells such as neutrophils and monocytes, and increased production of pro- and anti-inflammatory molecules.28,29 This robust response serves to promote repair, but it could be compromised by the competing influence of a peripheral infection over time. Therefore, we hypothesize that the immune stimulus of post-injury UTI complicates recovery and worsens outcome after TBI. Here, we establish a model of post-TBI UTI that remains localized to the urinary bladder in female mice. The effects of bladder infection after TBI are notable and include exacerbated cognitive impairment and elevated aspects of neuroinflammation compared to TBI alone. Together, these studies highlight the negative influence of bladder infection after TBI and support the need for continued investigation.
Materials and Methods
Subjects
Female C57Bl/6 mice (n = 97) from Charles River Laboratories (Wilmington, MA) were used for all experiments. Mice were single housed in The Ohio State University Laboratory Animal Resource facilities. Mice were given access to food and water ad libitum under controlled temperature (72–76°F) and humidity with a 12:12 h light cycle (lights on from 6:00 AM to 6:00 PM). Enrichment objects were provided to reduce the stress of single housing. At selected time points, mice were euthanized with CO2 asphyxiation, and tissues were harvested according to the experimental design. All animal conditions were in accordance with the principles and guidelines set forth by the National Institutes of Health’s Guidelines for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of The Ohio State University.
Experimental design
First, two doses of UPEC were examined following TBI. The primary goal of this experiment was to identify a dose of UPEC that would remain localized to the urinary bladder without spreading to the kidneys. Mice received either a moderate lateral fluid percussion injury (LFPI) or a sham injury. At 3 days post-injury (DPI), mice were inoculated transurethrally with either 1.0 × 106 colony-forming unit (CFU) or 1.0 × 107 CFU of the model strain UTI89. Urinalysis was performed at baseline 0 DPI (before injury), 4 DPI, 6 DPI, 10 DPI, and 17 DPI to confirm and examine the progression of the UTI. At the designated endpoints (4 DPI, 10 DPI, and 17 DPI), mice were euthanized by CO2 asphyxiation, kidneys and urinary bladders were harvested and cultured for E. coli. Animal numbers were n = 3 for TBI groups and n = 2 for Sham groups. One naïve control was added to assess the overall effect of surgery/injury and inoculation (Fig. 1A). Next, the behavioral and immune response to 1.0 × 106 CFU of UTI89 UPEC was determined after TBI. Separate cohorts of mice received either a moderate LFPI or a sham injury. At 3 DPI, mice were inoculated transurethrally with either vehicle (0.01 M phosphate-buffered saline [PBS]) or 1.0 × 106 CFU of UTI89 UPEC, resulting in a 2 (Sham, TBI) × 2 (Vehicle, UTI) factorial design. Urinalysis was performed at baseline 0 DPI (before injury), 4 DPI, 6DPI, and 10 DPI to confirm and examine the progression of the UTI. Y-maze was conducted 6 DPI, and the open field was performed 8 DPI. A subset of mice was euthanized by CO2 asphyxiation following Y-maze behavioral assessment 6 DPI to examine blood cytokines by enzyme-linked immunosorbent assay (ELISA), brain and blood immune cells using flow cytometry, and the presence of E. coli in the blood (n = 7–8 per experimental group). The remaining mice were euthanized by CO2 asphyxiation 10 DPI, and brains and urinary bladders were dissected and processed for immunofluorescent (IF) histology (n = 9 per experimental group, Fig. 1D).
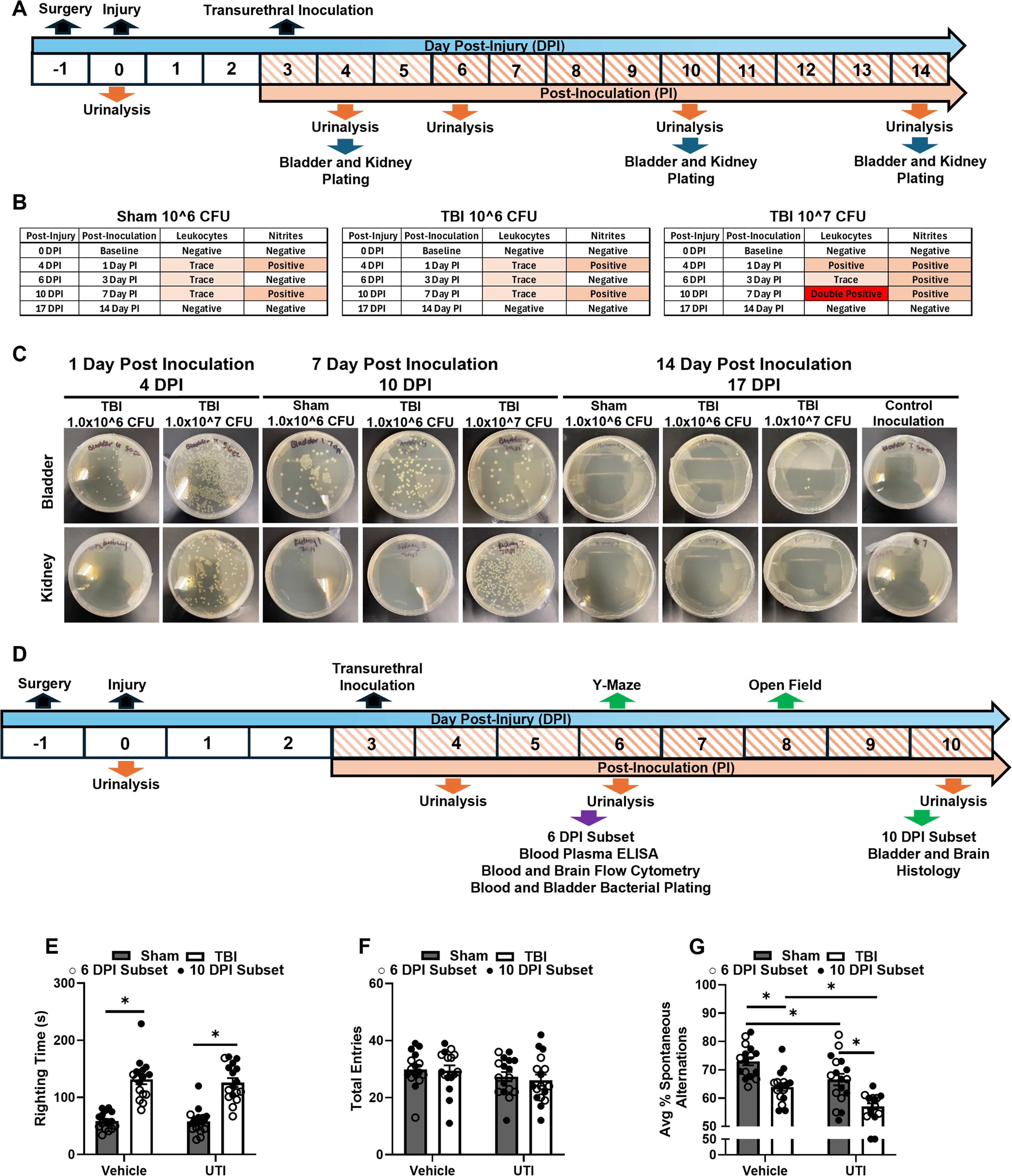
A localized dose of UTI89 Escherichia coli causes exaggerated cognitive deficits following TBI.
Lateral fluid percussion injury
All surgical and injury procedures were performed as previously described.30–33 Briefly, all mice were anesthetized with 4% isoflurane. The head was shaved prior to placement in a stereotaxic frame. Following sterilization of the skin, a midline incision was made to allow clear access to the skull surface. A 3 mm craniotomy was performed midway between bregma and lambda over the parietal lobe with the underlying dura remaining intact. A modified Luer-Lock syringe hub was attached using super glue and Ortho-Jet dental acrylic. The hub was then filled with 0.9% saline, and the mice were injected with 2 mL of 0.9% saline subcutaneously for rehydration before being placed back in their home cage. The following day, all mice were anesthetized with 4% isoflurane. For those in the TBI group, the hub was filled with 0.9% saline before attachment to the fluid percussion device (Custom Design and Fabrication, Richmond, VA). A weighted pendulum from a pre-set height was released, creating a fluid pulse (∼1 atm of pressure) onto the exposed intact dura and inducing a moderate severity injury. Following the injury, the hubs were removed, and the incision was closed with staples. For those in the Sham group, mice were anesthetized and attached to the fluid percussion injury device, but the pendulum was not released to induce injury. Incisions were immediately closed with staples. Injury severity was assessed by measuring latency to the self-righting reflex following both TBI and sham procedures (Fig. 1E).
UPEC strain UTI89 culturing
The model UPEC strain UTI89 was generously provided by the Hultgren laboratory and used to induce UTI as previously described. 34 A swab from the stab agar was placed in lysogeny broth (LB) and incubated statically overnight at 37°C and 5% CO2. About 1 mL of E. coli from the LB was then diluted in 25 mL of LB and again was incubated statically overnight at 37°C and 5% CO2. The second incubation was done to optimize expression of the extracellular attachment organelle called type 1 pili, which is critical for E. coli adherence and infection of the urinary bladder wall. 35 The E. coli was then centrifuged at 5000 rpm for 5 min at 4°C. The pellet was resuspended in 10 mL of 0.01 M PBS at pH 7.4, and the optical density of the E. coli was read on a spectrophotometer to determine the concentration of E. coli. From the optical densities, dilutions were calculated for a 1.0 × 106 CFU and 1.0 × 107 CFU concentrations. A yeast agglutination assay was performed to confirm robust expression of type 1 pili.
Transurethral inoculation
Under a biosafety cabinet, urinary catheters were made by placing PE10 tubing fully over a 30G 0.5-inch long blunt syringe needle and cutting ∼0.5″ from the tip of the needle. Catheters were placed in a glass petri dish and ultraviolet sterilized for 30 min. At the time of inoculation, the end of the catheter was trimmed to ∼1–2 mm from the end of the needle. Urinary bladders were fully emptied of urine prior to inoculation. Under a biosafety cabinet, mice were anesthetized with 4% isoflurane. Catheters were then placed on a 1 mL slip-tip tuberculin syringe, and 50 µL of E. coli or vehicle was drawn. Medical-grade lubricating jelly was added to the end of the catheter and placed in the urethral opening. The catheter was slowly inserted transurethrally up to the hilt of the syringe needle into the urinary bladder. Slow and constant pressure was applied to the catheter to inject the vehicle or E. coli transurethrally to ensure that it empties into the urinary bladder. Mice were allowed to recover from anesthesia before returning to their home cage.
Urinalysis
Urinary bladders were gently expressed, and the urine was collected into a small petri dish. Ten microliter of urine was placed on the leukocyte and nitrite squares of the urine test strips (Roche Diagnostics, Cat. No. 11895354160). Color change was compared to the chart supplied by the manufacturer.
Bacterial plating
Urinary bladders and kidneys were placed in glass mortars with LB broth for immediate homogenization and bacterial plating. Ten microliter of the homogenate or 20 μL of blood was added to LB agar plates and streaked using a glass spreader. Plates were sealed with parafilm and statically incubated overnight at 37°C with 5% CO2. Plates were visually assessed for the presence of bacterial colonies and growth (i.e., yes/no).
Y-maze
Y-maze was used to assess spatial working memory as previously described.31,33 Briefly, a mouse was placed in the maze and allowed to freely explore for 5 min. The maze was thoroughly cleaned with 70% ethanol between mice. The total number of arm entries and the total number of spontaneous alternations were scored. Data were analyzed as total arm entries and percent spontaneous alternations of total arm entries minus 2.
Open field
The open field was performed as previously described and used to assess motor and anxiety-like behavior.31,33 A mouse was placed facing the corner of a square plexiglass chamber and freely explored for 10 min. Each chamber was thoroughly sanitized with 70% ethanol between each animal tested. Total distance and time spent in the center and periphery were tracked and recorded by floor-level light beams in the maze along the edges of the chamber (San Diego Instruments). Outcome measures assessed were total center and peripheral time, total center and peripheral entries, total distance, and total resting time.
Enzyme-linked immunosorbent assay
A Meso Scale Diagnostics (MSD) V-Plex multi-spot assay system (MSD, Cat. No. K15048) was used to analyze blood cytokines (interferon-γ [IFN-γ], tumor necrosis factor-α [TNF-α], IL-1β, IL-6, and IL-10). Mice were euthanized via CO2 asphyxiation. Approximately 400 μL of blood was drawn from euthanized mice by transcardial puncture into centrifuge tubes containing EthyleneDiamineTetraacetic Acid (EDTA). Blood was centrifuged at 2000 g for 20 min at 40C, plasma was collected and stored at −80°C until the time of assay. Assay was performed according to the manufacturer’s protocol. The 96 well plates were read and analyzed on an MESO QuickPlex SQ 120MM microplate reader. Results were analyzed using MSD’s Discovery Workbench Software, which calculates the analyte’s concentration using a four-parameter logistic model with a 1/Y2 weighting.
Flow cytometry
Following CO2 asphyxiation, blood and the ipsilateral brain were rapidly collected 6 DPI. Blood was collected via cardiac puncture into EDTA-lined syringes and processed as previously described. 36 Briefly, red blood cells were lysed, remaining cells were pelleted, and supernatant was removed. Cell pellets were incubated with the following antibody solution: anti-CD16/CD32 antibody (Fc receptor blockage), Ly6G (FITC), Ly6C (PerCP-Cy5.5), CD11b (APC), CD3 (APC-Cy7), and B220 (PE-Cy7) for 15 min at room temperature. Cells were resuspended in PBS for analysis. For leukocyte isolation of the CD11b+ enriched layer, brain samples were homogenized, pelleted at 900 g for 6 min, and supernatant was removed. Cell pellets were resuspended in 70% isotonic Percoll. A discontinuous Percoll density gradient was then applied in three layers: 50%, 35%, and 0% (PBS) isotonic Percoll. The gradient was centrifuged for 20 min at 2070 g with low acceleration and brake. Previous studies have reported that viable cells isolated by Percoll density gradient yield >90% leukocytes.37,38 After centrifugation, the fat layer was removed from all tubes. The CD11b+ enriched layer was collected from the interphase between the 70% and 50% Percoll layers. After collection, the layer was washed to remove any remaining Percoll. After pelleting and supernatant removal, cells were incubated in the following antibody solution: Fc receptor block (anti-CD16/CD32), Ly6C (PE), Ly6G (FITC), CD45 (PerCP-Cy5.5), CD11b (APC), and B220 (PE-Cy7) for 15 min at room temperature. Samples were run using spectral flow cytometry on the Cytek Aurora 3-laser system. For spectral unmixing and autofluorescence removal, single stain reference controls were used. The fluorescence-minus-one method was used to assess nonspecific binding and confirm positive labeling for cell populations. Compensation was performed for brain samples using NXN plots on the Cytek Aurora flow cytometer. All data were analyzed using FlowJo software. In FlowJo, the gating strategy began with exclusion of debris from each sample, determined by forward scatter and side scatter. This was followed by the removal of doublets, creating a gate of all single cells. Cell populations of interest were then gated within the single cells gate, representing a percentage of all single cells. For most cell types, double positive populations were gated (CD11b+CD45Hi-Int, CD11b+Ly6C+, CD11b+Ly6CHi-Int, CD11b+Ly6G+). B cell populations include all B220+ cells gated against CD11b (both CD11b+B220+ and CD11b−B220+).
IF histochemistry
At 10 DPI, the remaining mice in each experimental group were euthanized by CO2 asphyxiation. Mice were perfused with 0.01 M PBS transcardially; brains were removed and post-fixed in 4% paraformaldehyde for 48 h and then stored in 30% sucrose for 48 h at 4°C. Urinary bladders were removed and post-fixed in 4% paraformaldehyde for 48 h and then stored in 30% sucrose for 48 h at 4°C.
Brains and urinary bladders were embedded in optimal cutting temperature compound, frozen on dry ice in ethanol, and stored at −20°C until the time of sectioning. Urinary bladders were sectioned at 15 µm directly on a slide with approximately 8 bladder sections per slide. Slides were stored at −20°C until the time of staining. A hydrophobic PAP pen was used to mark around the urinary bladder sections to perform on-slide IF histochemistry. Bladder sections were washed in 0.01 M PBS with 0.1% Triton X-100 (PBST) and then incubated in 5% normal donkey serum and 0.3% Triton X-100 in 0.01 M PBS for 2 h at room temperature. The bladder sections were then incubated overnight in primary antibody at 4°C: Ly6G (1:200; Abcam, Cat. No. ab210204), F4/80 (1:100; Serotec, Cat. No. MCA497), and CD11b (1:250; Serotec, Cat. No. MCA74G). The next day, the sections were washed with PBST and then incubated in a fluorescent conjugated secondary antibody for 1 h; for all donkey anti-rat 594 (1:1000; Invitrogen, Cat. No. A21209). Slides were then cover slipped using Fluoromount-G with DAPI. Ly6G, F4/80, and CD11b were imaged using a Nikon Eclipse Ti2 microscope at 20× magnification. Images were taken as a sequential z-stack every 1 µm, resulting in 12–15 images per stack. An enhanced depth of focus (EDF) image, which combines the focal points of all the z-stack images into one enhanced image, was used for quantification.
Mouse brains were then sectioned at 30 µm using a Leica CM1850 cryostat (Leica Biosystems). Brain sections were transferred to cryoprotectant and stored at −20°C until IF. Brain sections were first washed in 0.01 M PBS to thoroughly remove cryoprotectant and then washed in PBST to increase cellular permeability. To help reduce background, brain sections were blocked in 5% normal donkey serum and 0.3% Triton X-100 in 0.01 M PBS with constant rotation for 2 h at room temperature. The brain sections were then incubated overnight in primary antibody with constant rotation at 4°C; rabbit anti-Iba1 (1:500; Wako Chemicals, Cat. No. 019-19741), goat anti-GFAP (1:500 Abcam cat# 53554), and rat anti-CD68 (1:500; Abcam, Cat. No. 53444). Brain sections were washed with PBST and then incubated in a fluorescent conjugated secondary antibody with constant rotation for 1 h at room temperature; donkey anti-rabbit 647 for Iba1 (1:1000; Invitrogen, Cat. No. A31573), donkey anti-goat 594 for GFAP (1:1000; Invitrogen, Cat. No. A332758), and donkey anti-rat for CD68 (1:1000; Invitrogen, Cat. No. A21208). The brain sections were mounted and cover slipped using Fluoromount-G with DAPI (Fisher, Cat. No.501128966). Iba1 and GFAP were imaged with the same Nikon Eclipse Ti2 microscope at 20× magnification. Iba1 and CD68 co-label were imaged at 40× magnification. —Two to three images were taken for the regions of the hippocampus ipsilateral to the injury site and cortex adjacent to the lesion site within each animal. For the regions of the medial prefrontal cortex (mPFC), two to three images of both the ipsilateral and contralateral sides were taken. Images were taken as a sequential z-stack every 1 µm, resulting in 13–16 images per stack. EDF images were taken for each sequential z-stack and used for quantification. Brain regions of interest were the hippocampal CA1, CA3, and dentate gyrus, the retrosplenial and sensorimotor cortex near the site of injury, and the mPFC, including the cingulate cortex, prelimbic cortex, and infralimbic cortex.
Image quantification
Quantification for all IF of the mouse brains and urinary bladders was performed using FIJI. For the urinary bladder tissue, four EDF images, spaced apart in four quadrants, were taken per urinary bladder section and used for quantification. A blinded investigator calculated the percent area of Ly6G, F4/80, and CD11b by first subtracting background and then by manually thresholding to consistently capture the stained areas. The four quadrants per bladder section were averaged together and then averaged with two other sections of the same bladder for the final percent area. For brain tissue, an EDF image for each region of interest within each section was used for quantification. A blinded investigator calculated percent area of Iba1 and GFAP by first subtracting the background and then by manually thresholding to consistently capture the microglial somas and processes and the astrocytic somas and branches. For the regions of the hippocampus, retrosplenial and sensorimotor cortex, —two to three brain sections from each subject were quantified and averaged for the final percent area. For the regions of the mPFC, —two to three sections from the ipsilateral and contralateral were quantified. Both sides were averaged independently and then combined and averaged for the final percent area. For CD68/Iba1 colocalization, CD68 and Iba1 images were manually thresholded separately, and binary masks of the positive stain for each were overlayed using the FIJI image calculator. This created a single mask representing portions of the image that were positive for both CD68 and Iba1. Percent area was then calculated and reported.
Statistics
A two-way analysis of variance was performed in GraphPad Prism 10 to compare changes in behavioral metrics, IF, ELISA, and flow cytometry in Sham or TBI mice between vehicle and UTI89 UPEC-inoculated animals. Tukey’s HSD post hoc test was performed to compare individual group means when main effects and interactions were statistically significant (α set at 0.05). All comparisons with p ≤ 0.05 are reported and displayed within the graphs and tables.
Results
1.0 × 106 CFU UTI89 remains localized to the urinary bladder after TBI
We sought to identify a bacterial dose sufficient to induce UTI while remaining localized to the urinary bladder without migration to the kidneys, causing pyelonephritis to recapitulate the most common UTI. Mice received either Sham or TBI, and then 3 DPI received either 1.0 × 106 or 1.0 × 107 CFU of the model UPEC strain UTI89 (Fig. 1A). Urinalysis was performed throughout the time course to identify the presence of leukocytes and nitrites, which are markers of an immune response to UTI and are commonly used in clinical settings to diagnose UTIs. As expected, no mice showed any presence of leukocytes and nitrites in the urine at baseline (before injury, 0 DPI). Sham and TBI mice receiving 1.0 × 106 CFUs had trace presence of leukocytes in the urine 1, 3, and 7 days after inoculation, as well as nitrites at 1 and 7 days after inoculation (4 and 10 DPI, respectively). TBI mice receiving 1.0 × 107 CFUs had a positive presence of leukocytes in the urine at 1 and 3 days after inoculation (4 and 6 DPI, respectively) with a double positive reading at day 14 or 17 DPI, while days 1, 3, and 7 (4, 6, and 10 DPI, respectively) were positive for nitrites in the urine, as expected given that inoculum dose of UTI89 can mediate infection outcome. 39 By day 14 post inoculation (17 DPI), all mice had negative readings for both leukocytes and nitrites in the urine, suggesting the infection cleared, as is expected for a single UTI89 infection in C57BL/6 mice 40 (Fig. 1B).
To measure bacterial colonization, urinary bladders and kidneys were harvested, homogenized, and plated on LB agar at 1, 7, and 14 days post inoculation (4, 10, and 17 DPI, respectively). At 1 and 7 days post inoculation (4 and 10 DPI, respectively), 1.0 × 106 and 1.0 × 107 CFU showed bacterial colonization within the urinary bladder, with the higher dose having more bacterial colonies extensively. Notably, only 107 CFU caused bacterial colonization in the kidneys, and this was apparent at 1 and 7 days post inoculation (4 and 10 DPI, respectively). By day 14, there was no bacterial presence in the urinary bladder or kidney in either dose, indicating resolution of the infection, which is expected in C57Bl/6 mice. 39 Together, these data show that the lower dose of 1.0 × 106 CFU remained localized to the urinary bladder, and that bladder colonization was similar in TBI- and sham-treated mice (Fig. 1C).
Transurethral UPEC inoculation 3 DPI increases leukocytes and nitrites in the urine independent of TBI
Our next objective was to define the behavioral and immune effects of UTI after TBI using 1.0 × 106 CFU UTI89 (Fig. 1D). Mice received either Sham or TBI. Latency to regain righting reflex (righting time) was taken as a measure of injury severity. As expected, TBI increased righting time, main effect of injury, F(1, 63) = 113.6, p < 0.0001, indicating that any subsequent differences between Sham and TBI groups were due to UTI (Fig. 1E). Next, half of the mice in each experimental group received vehicle inoculation, and the other half received 1.0 × 106 CFU of UPEC 3 DPI. Urinalysis was performed as previously described. At baseline (0 DPI), both Sham and TBI mice were negative for leukocytes and nitrites in the urine. Sham and TBI mice receiving UTI89 inoculation showed trace presence of leukocytes in the urine throughout the time course and were positive for nitrites 4 and 10 DPI, replicating previous results (Fig. 1B). Together, the urinalysis indicates the presence of the infection in UPEC-inoculated mice through the time course of the study, which ended 10 DPI.
Post-injury UTI exaggerates TBI-induced cognitive deficits 6 DPI
The Y-maze was used to assess spatial working memory 6 DPI. There were no differences in total arm entries between experimental groups (Fig. 1F), suggesting no gross motor deficits. Main effects of injury, F (1, 61) = 36.14, p < 0.0001, and infection, F(1, 61) = 18.52, p < 0.0001, were detected in the percentage of spontaneous alternations, indicating that both TBI and UTI influenced spatial working memory (Fig. 1G). Post hoc comparisons confirmed that TBI reduced the percentage of spontaneous alternations compared to Sham (p = 0.0006). Sham UTI also significantly decreased spontaneous alternations compared to Sham (p = 0.0205). TBI UTI reduced the percentage of spontaneous alternations compared to Sham UTI (p = 0.0003); and strikingly, the combination of TBI and UTI exaggerated the impairment in spontaneous alternations compared to TBI alone (p = 0.0152).
UTI89 is not detected in the blood 6 DPI
To determine if E. coli escaped the bladder and entered circulation at the time of Y-maze assessment, urinary bladders and blood were collected immediately after behavioral testing in a subset of mice 6 DPI. Homogenized bladders and blood were separately plated onto LB agar plates (Fig. 2A). After a 24-h incubation period, the presence of bacteria was assessed. As expected, UTI89 was only present in the bladders of UPEC-inoculated mice. There was no UTI89 detected in the blood of any experimental group. Therefore, these results provide evidence that UTI89 remains localized to the urinary bladder in this model, and that any peripheral immune response 6 DPI was not influenced by ongoing bacterial sepsis.
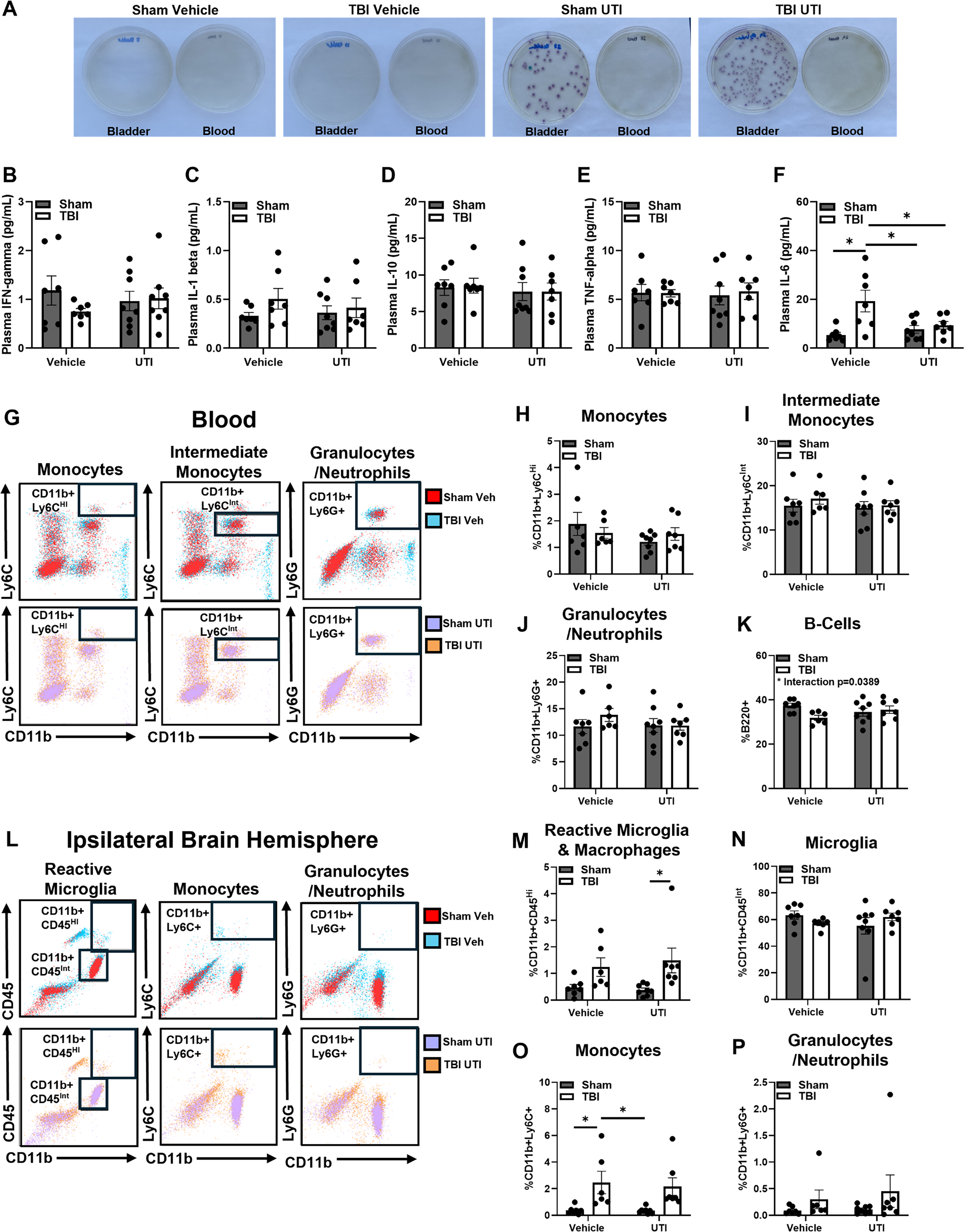
UTI after TBI attenuates plasma IL-6 without amplifying peripheral immune response 6 DPI:
UTI after TBI attenuates plasma IL-6 6 DPI
We then sought to explore the potential influence of circulating cytokines in coordinating a bladder–brain response to post-TBI UTI. Blood plasma was collected in the same subset of mice 6 DPI used in Figure 2A. Key cytokines were examined through a multiplex assay, including IFN-γ, TNF-α, IL-1β, IL-10, and IL-6. No significant changes were observed in plasma IFN-γ, TNF-α, IL-1β, and IL-10 following TBI and UTI (Fig. 2B–E). A main effect of TBI was detected in plasma IL-6, F(1, 25) = 9.869, p = 0.0043, and post hoc analysis revealed a significant increase in TBI compared to Sham (p = 0.0034), Sham UTI (p = 0.0121), and TBI UTI (p = 0.0456). Unexpectedly, UTI after TBI muted the plasma IL-6 response 6 DPI (Fig. 2F). C57BL/6 mice that are infected with a single dose of UTI89 do not have a marked systemic immune response at 24-h post inoculation, 40 although there is a robust local immune response in the bladder, which subsides at 48-h post infection. 41 These data suggest that a pronounced immune response in this model may have been elicited during early infection but then subsided by 6 DPI.
Peripheral immune response to TBI is not exacerbated by UTI 6 DPI
Both TBI and UTI elicit a peripheral immune response,40,42,43 which could bridge signaling between the injured brain and infected bladder. Therefore, flow cytometry was performed in the same subset of mice 6 DPI to assess the effects of TBI and UTI on the distribution of peripheral immune cell populations. Blood and the ipsilateral brain hemispheres were collected for evaluation. There were no significant differences in the distribution of monocytes (%CD11b+LyCHi, %CD11b+Ly6CInt) and granulocytes (%CD11b+LyCG+) in blood (Fig. 2H–J). However, there was an interaction effect within %B220+ B cells in the blood, F(1, 24) = 4.771, p < 0.05, Figure 2K. In the brain, a main effect of injury was detected in the %CD11b+CD45Hi population, F(1, 24) = 10.81 p < 0.05, which contains highly reactive microglia and peripheral macrophages. Post hoc comparisons confirmed a significant difference between TBI and Sham mice (p = 0.0411; Fig. 2M). However, there were no significant differences in %CD11b+CD45Int microglia between any of the groups (Fig. 2N). A main effect of TBI was detected in %CD11b+LyCHi monocytes, F(1, 24) = 15.72, p < 0.05, and post hoc comparisons confirmed an increase in TBI compared to Sham (p = 0.0368) and Sham UTI (p = 0.0283; Fig. 2O). There were no significant differences in the infiltration of granulocytes (%CD11b+LyCG+) into the ipsilateral hemisphere (Fig. 2P). These findings support previous studies demonstrating that TBI elicits a peripheral immune response resulting in brain infiltration during the subacute phase of recovery, but this response is not exaggerated by UTI.
TBI and UTI do not alter open-field behavior 8 DPI
Open field was used to assess anxiety-like behavior and overall activity 8 DPI. No significant differences were detected between experimental groups in any metric of the open-field behavioral test (center and peripheral entries, center and peripheral time, total distance, and resting time (Fig. 3A–F). These results demonstrate that mice exhibited no anxiety-like behavior, motor deficits, or sickness behaviors due to injury or infection.
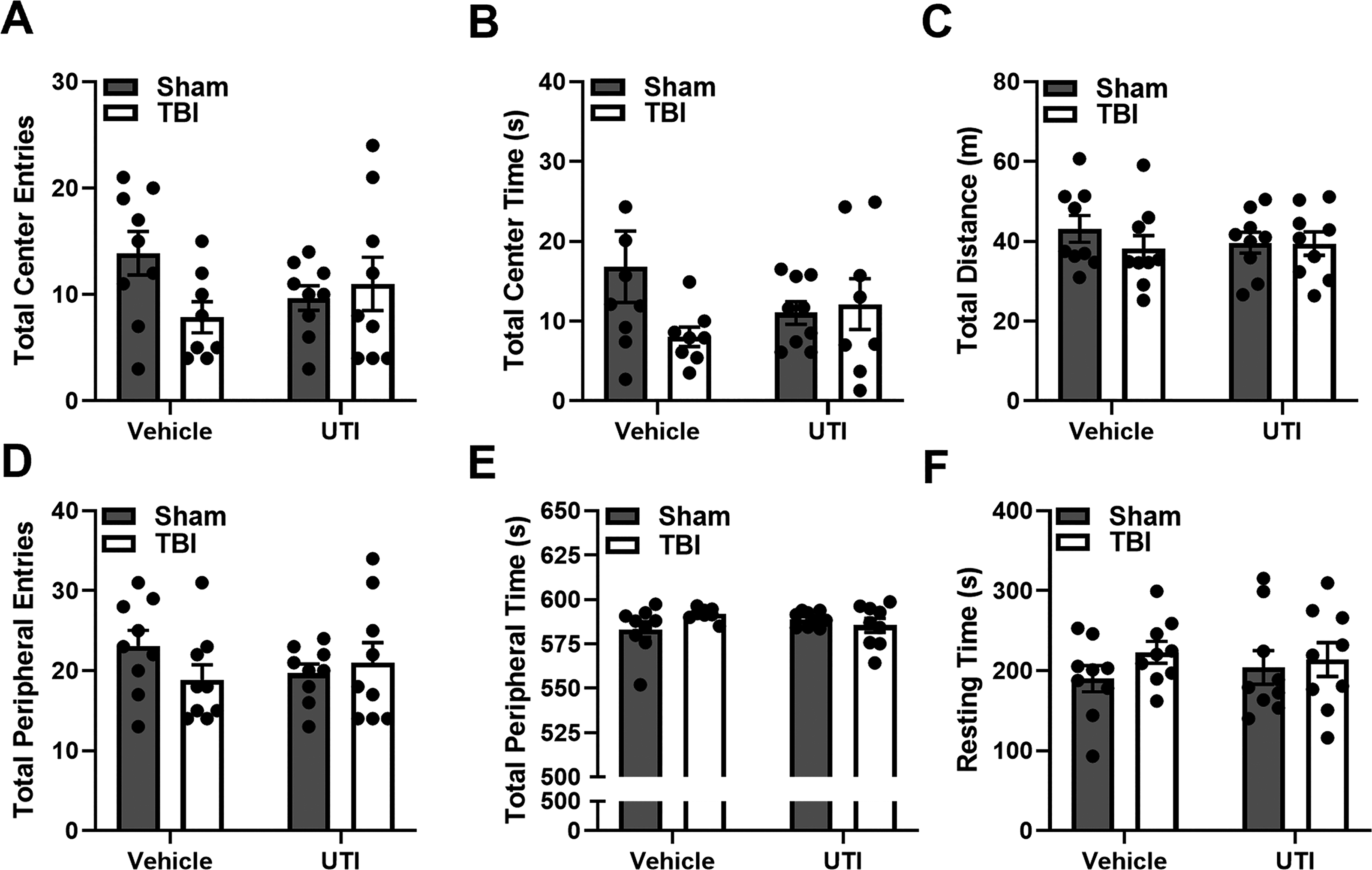
TBI and UTI do not alter open field behavior 8 DPI: Mice showed no observable differences in all open-field metrics, suggesting that there was no decreased activity due to motor deficits or UTI infection. Data presented as mean ± SEM. *p ≤ 0.05. Post hoc comparisons are visualized on the bar graphs. DPI, days post inoculation; SEM, standard error of the mean; TBI, traumatic brain injury; UTI, urinary tract infection.
TBI and UTI increase immune cell expression in the urinary bladder 10 DPI
Next, IF labeling of the urinary bladder 10 DPI was used to identify key immune cells altered in response to TBI and UTI, particularly innate immune cells by targeting CD11b (macrophages and neutrophils), Ly6G (granulocytes, including neutrophils), and F4/80 (macrophages; Fig. 4A). A main effect of injury was identified in percent area of CD11b expression, F(1, 20) = 11.35, p = 0.0031; Figure 4B. Post hoc comparisons revealed that CD11b expression was significantly elevated in TBI UTI mice compared to Sham and Sham UTI mice. An interaction effect, F(1, 20) = 4.774, p = 0.0410, and a main effect of injury, F(1, 20) = 11.35, p = 0.0004, was detected in Ly6G expression (Fig. 4C). Sham mice receiving UTI expressed less Ly6G than Sham and TBI mice (p = 0.0377 and p = 0.0014, respectively). However, TBI UTI mice showed significantly more Ly6G than Sham UTI mice (p = 0.0010). Finally, a main effect of injury was detected in percent area of F4/80 expression, F(1, 20) = 31.35, p < 0.0001; Fig. 4D. TBI alone caused significant increases in F4/80 compared to Sham mice (p = 0.0111), while UTI following TBI significantly increased F4/80 expression compared to Sham and Sham UTI mice (p = 0.0214 and p = 0.0014, respectively). Taken together, these data show that TBI and UTI both alter the immune response in the bladder and interactions between the insults are complex.
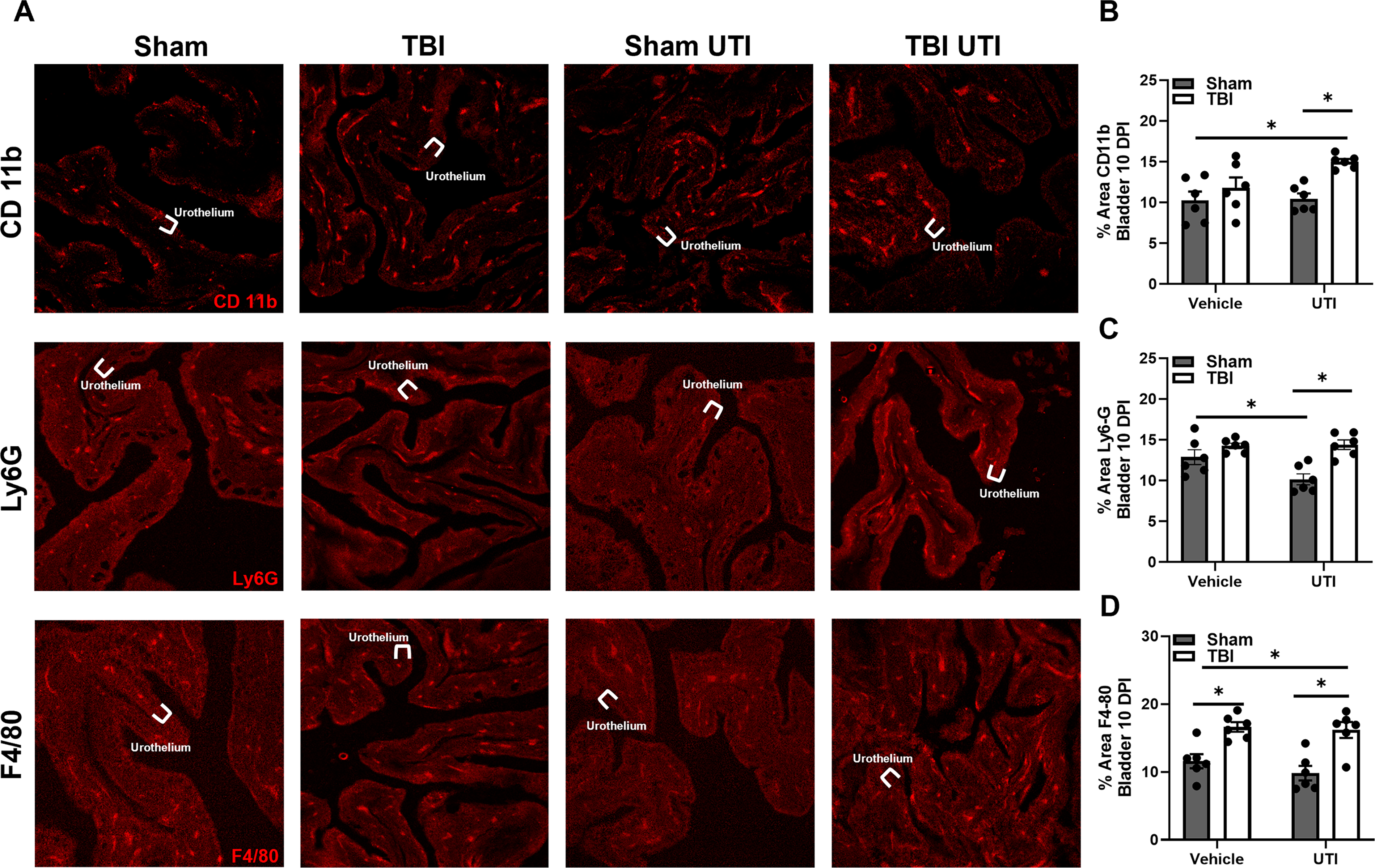
TBI and UTI increase immune cell expression in the urinary bladder 10 DPI.
TBI and UTI elevate GFAP expression in the hippocampus and cortex 10 DPI
Next, IF labeling was used to examine GFAP, a protein expressed by reactive astrocytes. Detailed statistical results can be found in Table 1. Main effects of injury and infection were identified in all brain regions examined, including CA1, CA3, dentate gyrus, retrosplenial cortex, sensorimotor cortex, cingulate cortex, prelimbic cortex, and infralimbic cortex (Figs. 5 and 6). However, these results must be contextualized to injury × UTI interaction effects that were also identified in most brain regions, including CA1, CA3, dentate gyrus, retrosplenial cortex, sensorimotor cortex, and infralimbic cortex. Because Sham UTI mice had higher GFAP than Sham vehicle controls in the hippocampus and cortical regions near the site of injury, the magnitude of the TBI-associated increase in GFAP was less pronounced in the presence of UTI. However, this trend did not extend to the cingulate or prelimbic cortices. Collectively, the data emphasize that injury and UTI do not act independently on GFAP expression. Yet, it is also possible that GFAP has reached a ceiling effect due to TBI such that TBI plus UTI can no longer enhance it in certain brain regions.

TBI and UTI elevate GFAP expression in the hippocampus and cortex at 10 DPI.
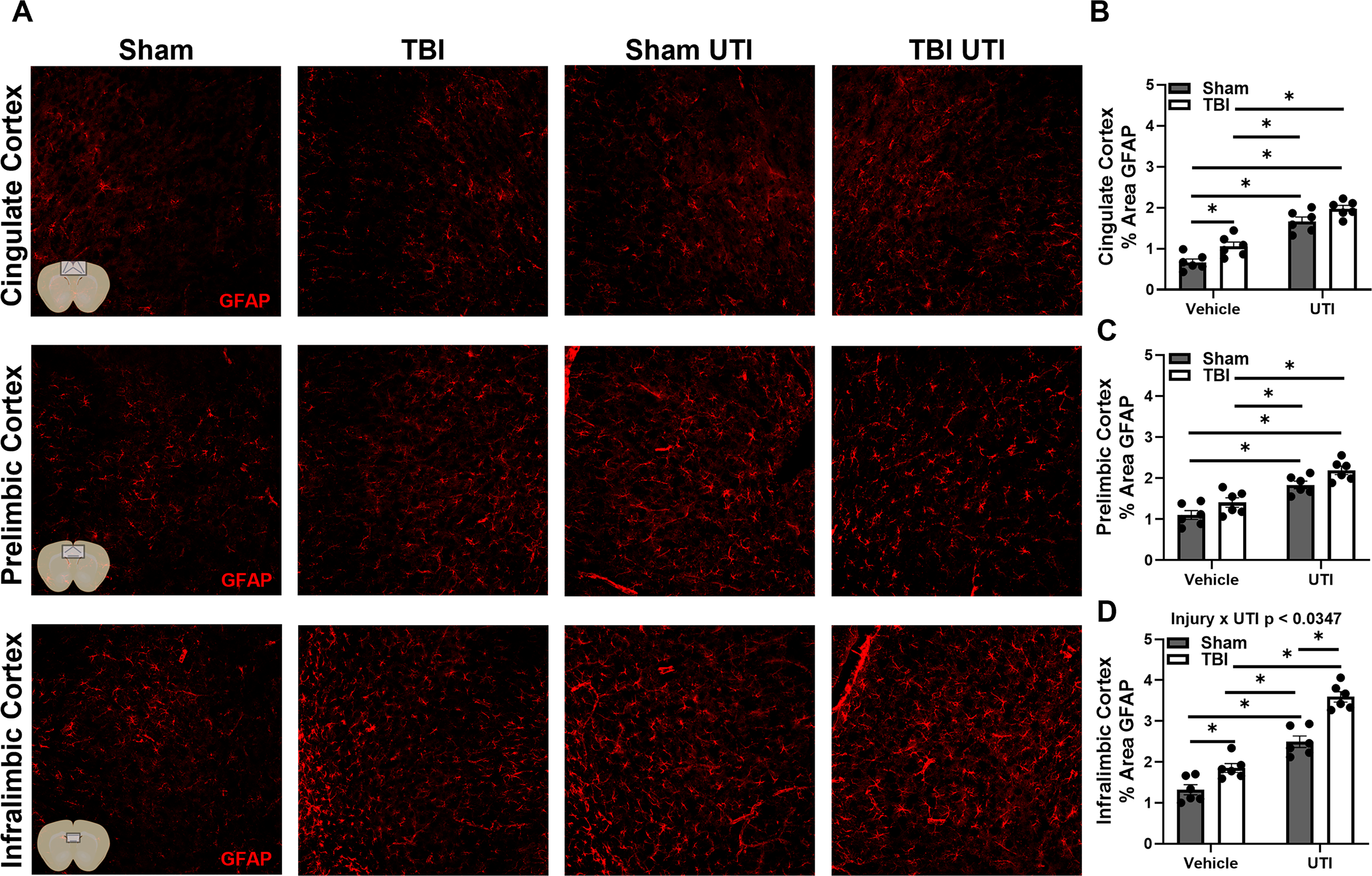
UTI exacerbates GFAP expression in regions of the mPFC.
Brain Immunofluorescent Histology for GFAP
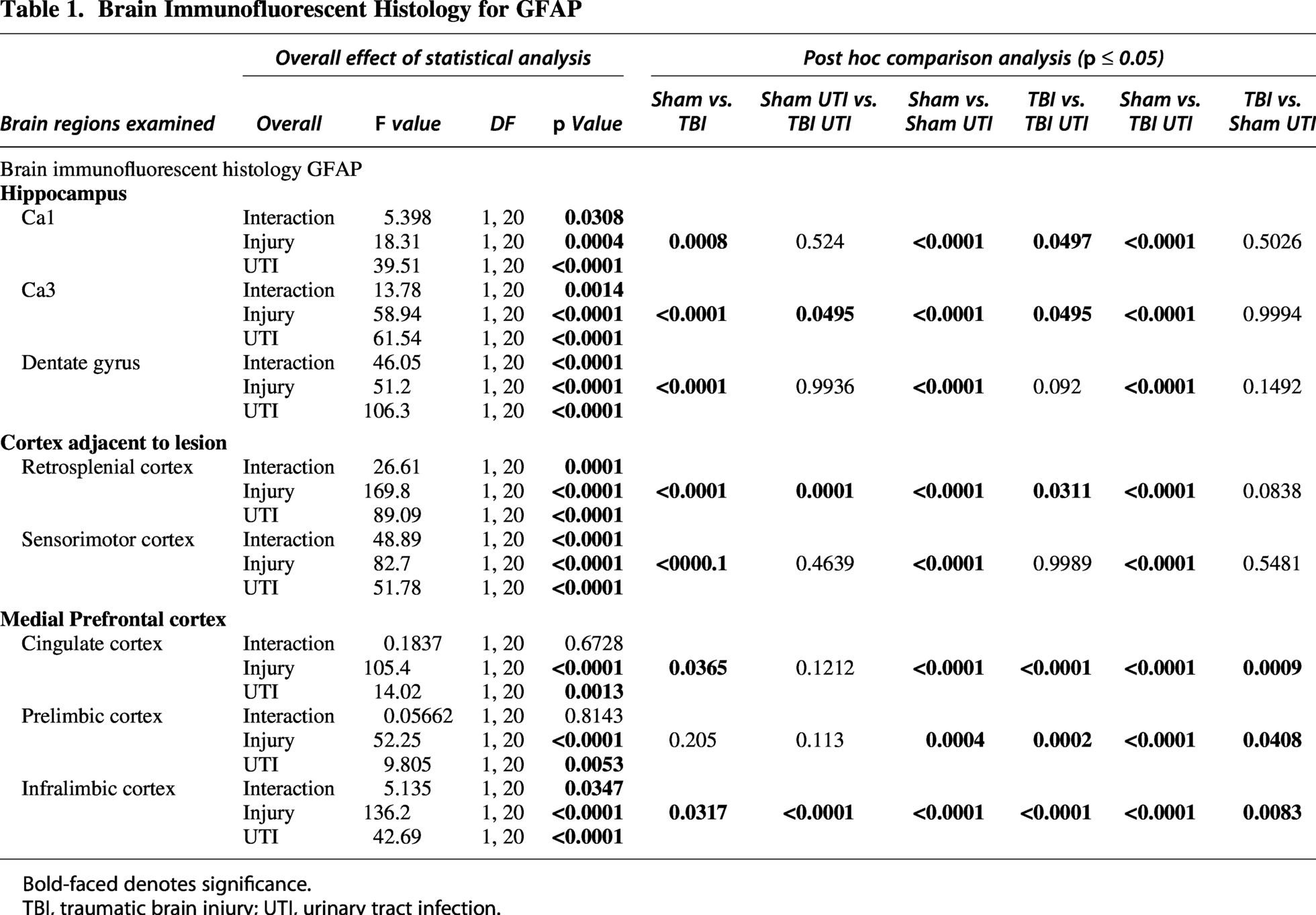
Bold-faced denotes significance.
TBI, traumatic brain injury; UTI, urinary tract infection.
UTI following TBI modifies Iba1 expression in a region-dependent manner 10 DPI
Next, we examined Iba1, which is highly expressed by reactive microglia. Detailed statistical results can be found in Table 2. Broadly, the Iba1 and GFAP results are consistent with main effects and interaction patterns, although localized to different brain regions. Main effects of injury and infection were identified in percent area of Iba1 in all brain regions examined, including CA1, CA3, dentate gyrus, retrosplenial cortex, sensorimotor cortex, cingulate cortex, prelimbic cortex, and infralimbic cortex (Figs. 7 and 8). The presence of injury × UTI interaction effects in the dentate gyrus, retrosplenial cortex, sensorimotor cortex, and infralimbic cortex underscores the need to consider these main effects within the context of their combined influence. Together, these results show that the Iba1 response reflects both additive and conditional effects of TBI and UTI.

UTI following TBI modifies Iba1 expression in the hippocampus and in the regions of the cortex adjacent to the lesion site.
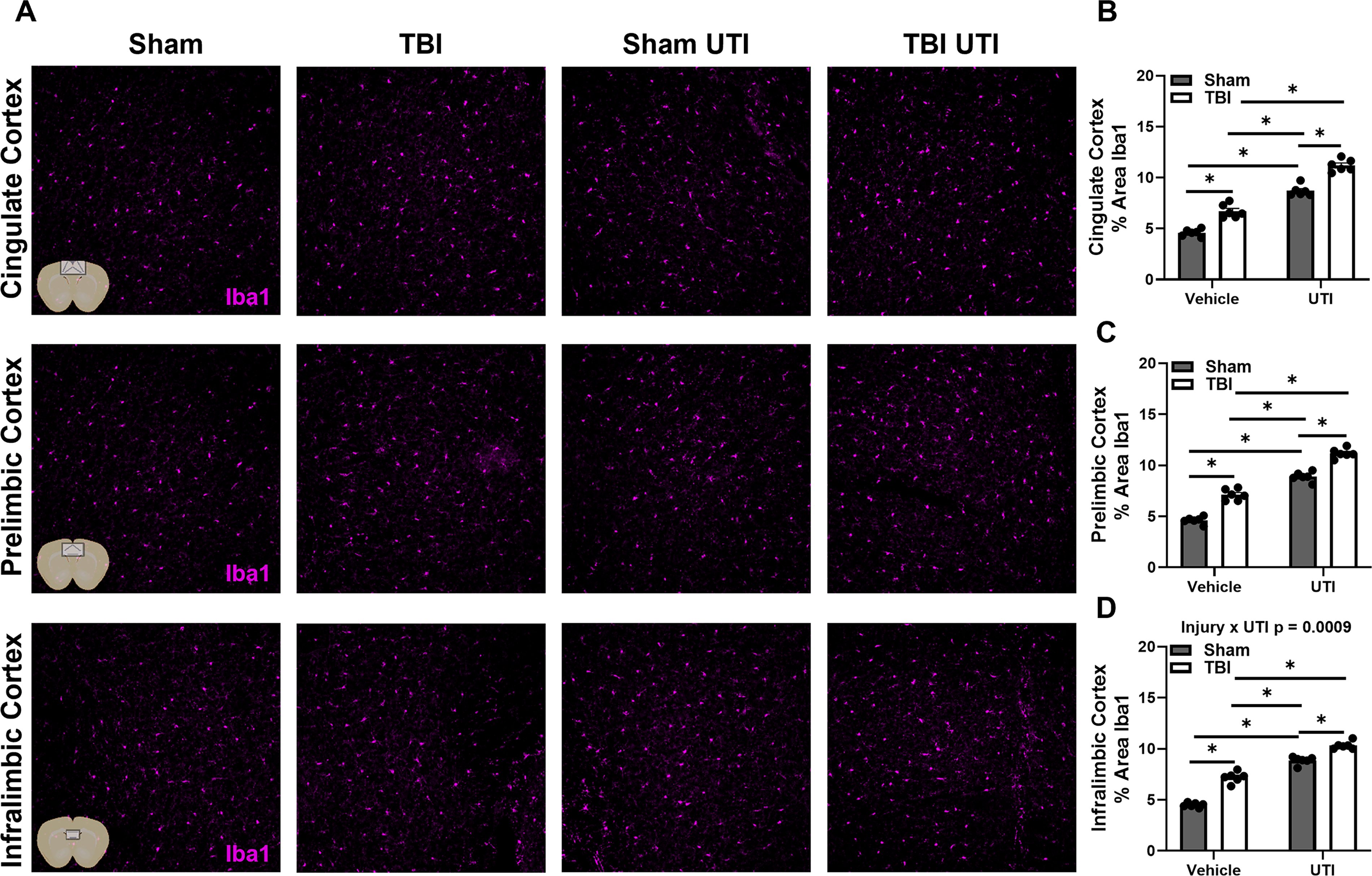
UTI after TBI exacerbates Iba1 expression in regions of the mPFC.
Brain Immunofluorescent Histology for Iba1
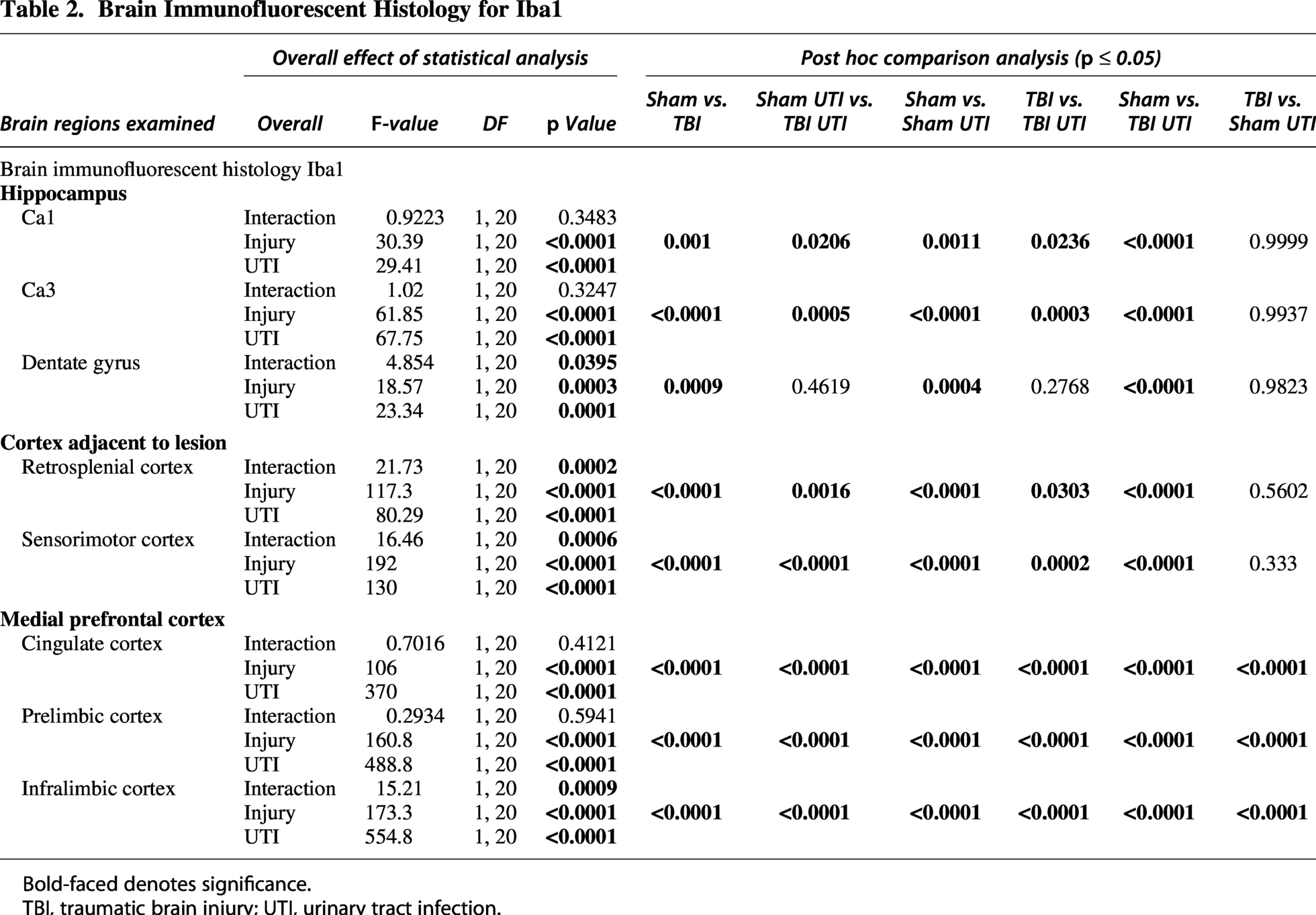
Bold-faced denotes significance.
TBI, traumatic brain injury; UTI, urinary tract infection.
UTI after TBI elevates microglial CD68 expression in regions of the hippocampus and cortex 10 DPI
Finally, to better define TBI- and/or UTI-induced changes in microglial activity, we examined CD68 colocalization with Iba1. CD68 is a transmembrane protein that is involved in the clearance of cellular debris and phagocytosis. Overall, TBI increased CD68/Iba1 expression in the dentate gyrus and all three regions of the mPFC, dentate gyrus: F(1, 20) = 4.240, p = 0.05; cingulate cortex: F(1, 20) = 4.716, p = 0.0421; prelimbic cortex, F(1, 20) = 0.0002; infralimbic cortex, F(1, 20) = 14.70, p = 0.001. UTI alone caused elevated CD68/Iba1 colocalization in the retrosplenial and infralimbic cortex, retrosplenial cortex: F(1, 20) = 9.386, p = 0.0061, and infralimbic cortex: F(1, 20) = 4.346, p = 0.05. Post hoc analysis revealed that TBI mice had higher CD68/Iba1 expression compared to Sham mice only in the infralimbic cortex (p = 0.0455). Also, TBI UTI mice had higher CD68/Iba1 compared to Sham UTI mice in the prelimbic cortex (p = 0.0016). UTI following TBI elevated CD68/Iba1expression in the dentate gyrus and retrosplenial, prelimbic, and infralimbic cortices compared to Sham mice (dentate gyrus; p = 0.05; retrosplenial cortex; p = 0.0368; prelimbic cortex; p = 0.0034; infralimbic cortex; p = 0.0024; Fig. 9A–F). No statistically significant differences were identified in CD68/Iba colocalization in the sensorimotor cortex, CA1, or CA3 (data not shown). These data reveal that microglia function is distinctly shaped by TBI and UTI, with their combination enhancing region-specific phagocytosis.

UTI following TBI elevates microglial CD68 expression in regions of the hippocampus and cortex.
Discussion
UTI is one of the most common infections worldwide and can affect TBI survivors. The paucity of research defining the pathophysiological and behavioral effects of post-TBI UTI calls for attention due to the severity of outcome in this comorbidity. Clinical studies show that premenopausal females are 30× more likely to get a UTI compared to age-matched males.6,44,45 Therefore, females experiencing uncomplicated UTI after TBI may be at enhanced risk for residual impairment compared to males, which have been the predominant group studied for TBI outcomes. To address this critical topic, we developed a model to study post-injury UTI in 3-month-old C57BL6 female mice using UPEC, the most common causative agent of uncomplicated UTI. We provide evidence that a transurethral inoculation of 1.0 × 106 CFU of the model UPEC strain UTI89 is sufficient to colonize the bladder for 7 days without traveling to the kidney and is no longer detected at 14 days post inoculation, reflecting key features of lower UTI. As expected, transurethral inoculation of 1.0 × 106 CFU of UPEC 3 DPI elevates leukocytes and nitrites in the urine of Sham and TBI mice. UTI exaggerates spatial working memory impairment in TBI mice compared to UTI-shams and TBI control mice 3 days after inoculation, providing evidence that peripheral infection specifically in the bladder can worsen distinct brain functions. Compellingly, UTI-induced impaired cognition occurs in conjunction with exaggerated GFAP and Iba1 percent area in multiple cortical and hippocampal regions in TBI mice compared to controls, implicating the host immune response as a potential mechanism for UTI-induced cognitive deficiencies. TBI increased percent area of CD68/Iba1 colocalization, and UTI after TBI enhanced this response in regions of the mPFC. Null results refine our understanding of UTI after TBI by showing that spatial memory deficits and alterations in brain IF arise without concomitant amplification of peripheral immune cell circulation or brain infiltration. Although a robust behavioral and neuroimmune response to UTI in Sham mice was initially unexpected, these results are consistent with the UTI model and highlight predictive nuance when combined with neurotrauma. Together, these main findings suggest that UTI aggravates TBI-induced glial reactivity and further impairs cognition after injury. These results emphasize the seriousness of post-injury UTI and highlight the importance of a bladder–brain axis in mediating post-injury neuropathology.
Well-characterized mouse models have been instrumental in elucidating molecular and cellular mechanisms that drive UTI pathogenesis, and this pre-clinical model has been used to predict and confirm aspects of UTI that are highly relevant to human pathophysiology (see review 46 ). Here, we examined two doses of UPEC strain UTI89 including 1.0 × 106 CFU and 1.0 × 107 CFU after TBI to determine bacterial presence in harvested bladders and kidneys. Only the 1.0 × 106 CFU dose remained localized to the bladder after injury, and no bacteria were seen in the kidneys of infected mice. This dosage is lower than that used in UTI-only studies, which is primarily 1.0 × 107–1.0 × 109 CFU. To be consistent with clinical practice, urinalysis was also used to confirm UTI. Detection of leukocytes and nitrites was similar between Sham and TBI mice, which strongly indicates UTI. Currently, we do not know why nitrites are negative in urinalysis 6 DPI. Although unexpected, this result consistently appeared in every single cohort. Further investigation is required to determine the biological significance of this observation. Additionally, urinalysis does not identify specific inflammatory cells that are activated within the urinary tract following inoculation. There is a well-established literature on mechanisms of UTI, including the immune response to UPEC (see review 47 ). It is worth highlighting that TBI increased expression of CD11b+, Ly6G+, and F4/80+ cells in the urinary bladder 10 DPI. The additive effects of UTI after TBI were subtle 10 DPI; however, this was 7 days after bacterial inoculation. Altogether, the results emphasize bladder vulnerability to inflammation for more than a week after TBI. Future studies can be designed to determine if TBI alters bladder histology, as well as the acute bladder response to infection.
In agreement with our hypothesis, UTI after TBI exaggerated spatial working memory deficits 6 DPI. To begin to understand signaling pathways linking bladder to brain, we performed a multiplex analysis of key plasma cytokines 6 DPI: IFN-γ, TNF-α, IL-1β, IL-10, and IL-6. Compellingly, IL-6 was the only cytokine influenced by the combination of TBI and UTI. Alone, TBI caused a significant elevation in plasma IL-6 6 DPI. UTI after TBI dramatically muted this response, suggesting that UTI may also be inducing an anti-inflammatory program during clearance of UPEC from the bladder. Notably, there were no significant differences in plasma IL-6 between Sham, Sham UTI, and TBI UTI groups. The absence of elevated plasma IL-6 at the time of behavioral testing suggests that the TBI UTI phenotype is not driven by ongoing IL-6 signaling in circulation 6 DPI. However, because cytokine signaling is dynamic, we cannot exclude the possibility that earlier IL-6 activity, or the activity of other inflammatory mediators, played a role in initiating or modulating the behavioral phenotypes or neuroimmune differences identified at 6 and 10 DPI, respectively. Previous work demonstrates that brain and blood IL-6 cause cognitive deficits following UTI. 48 However, the dosage of E. coli was 100 times higher (i.e., 1 × 108 CFU) than the one used in the present study. Additionally, evidence of bacteria was detected in the bladder, kidneys, and blood. 48 Bacteria were not detected in the blood following Sham or TBI in the present study. UTI of milder severity, such as the one we used in these studies, may influence the brain through alternative pathways. Nonetheless, these results justify further studies to determine the temporal and tissue-specific IL-6 response to post-injury UTI. Moreover, these observations suggest the possibility of a localized peripheral immune response in the bladder following UTI. While we would expect transurethral inoculation with UTI89 to induce a swift peripheral immune response (e.g., within hours), the time course of this response may be different in Sham and TBI mice. The bladder response to UTI89 is of interest for future studies.
Next, we utilized flow cytometry to define changes in peripheral immune cell circulation and infiltration to the brain 6 DPI. First, an unexpected interaction effect was observed in B cells, but there were no statistically significant differences in monocytes or neutrophils in the blood 6 DPI. Given that bacteria were not detected in circulation, we infer that direct microbial involvement is not elevating peripheral immune cell trafficking 6 DPI. Second, TBI increased the presence of reactive microglia and macrophages, as well as monocytes in the injured brain hemisphere, but this was not further affected by UTI. Therefore, UTI did not exaggerate peripheral immune cell infiltration to the brain 6 DPI. These results provide a snapshot of the peripheral immune response and do not exclude the possibility of altered trafficking or brain infiltration at other time points or via alternative pathways, which should be explored in follow-up studies.
The mPFC is distal to the site of brain injury, but it is required for successful completion of the Y-maze.5,49 Thus, we investigated the response of glia to TBI and UTI in the mPFC, alongside hippocampal and cortical regions near the epicenter of the injury. TBI alone increased GFAP and Iba1 in the mPFC compared to sham injury, and UTI inflated this response. Post hoc comparisons confirmed that the Iba1 percent area was highest in TBI UTI mice in all areas of the mPFC. Overall, similar trends in GFAP and Iba1 percent area were detected between experimental groups in cortical and hippocampal regions near the site of injury. However, the statistical patterns reveal that GFAP is frequently modified by the combination of injury and infection, and Iba1 is more often influenced by independent main effects of injury and infection. Together, this suggests that the neuroimmune effects of post-injury UTI vary by brain region and cell type. CD68/Iba1 colocalization provided insight into microglial phagocytic activity. As expected, brain injury increased the expression of CD68/Iba1 colocalization near the site of injury. However, the effect of injury on the expression of CD68/Iba1 colocalization was also apparent in the mPFC. UTI after TBI resulted in the highest expression of CD68/Iba1 colocations in many brain regions, but no statistically significant differences were detected between TBI and TBI UTI groups. Together, these results demonstrate that UTI after TBI worsens cognitive impairment, elevates neuroinflammation, and aggravates microglial reactivity in a region-dependent manner.
Previous pre-clinical work from our group shows that TBI-induced microglia priming is a gateway for post-injury immune insults to worsen long-term outcome, specifically cognition.28,50–52 Primed microglia have a dystrophic morphology with increased expression of Iba1 and CD68, antigen presentation molecules such as major histocompatibility complex II (MHCII), Toll-like receptors (TLRs), pro-inflammatory cytokines (IL-1β), and reduced expression of homeostatic molecules (CX3CR1).53–57 When these primed microglia are exposed to a peripheral immune challenge with lipopolysaccharide (LPS) 30 DPI, they mount an amplified inflammatory reaction that is associated with depressive-like behavior 43 and impaired memory consolidation. 28 Here, we extend these studies by examining the behavioral and immune response to a translationally relevant peripheral infection that commonly occurs in TBI patients.
Compellingly, LPS is a major structural component of the outer membrane of UPEC and plays a crucial role in mediating TLR4-dependent immune signaling within the bladder during infection.58–61 The innate immune response to UTI includes activation of neutrophils, monocytes, mast cells, and natural killer cells through pattern recognition receptors such as TLR (as reviewed10,11). Increased production and release of pro- and anti-inflammatory cytokines in response to UTI facilitate phagocytosis of pathogens, activate the complement system, and promote chemotaxis to the infection site. However, during this response, bacteria may exploit several mechanisms to invade and influence the central nervous system. This includes direct adhesion to endothelial or epithelial cells, paracellular entry through damaged barriers such as the BBB, and/or transmigration with infected peripheral immune cells. 62 Consequently, the behavioral and neuroimmune results presented here may result from the interplay of several distinct signaling pathways and therefore require additional investigation. While we highlight the role of microglia in this study, it is premature to exclude the potential role of other cellular players in the behaviors and neuroimmune changes reported here.
In terms of clinical applicability, UPEC offers several advantages over LPS in pre-clinical studies (see reviews63,64) as UPEC is a living, replicating organism. LPS is a molecule, serving as one antigen of UPEC that can cause an immune response through recognition by TLR4, which activates innate immune responses. UPEC engages the host through multiple pattern-recognition receptors. UPEC uses pili, specifically type 1 pili, to attach to the urothelium and secrete toxins such as hemolysin A. By inducing a true infection, UPEC has the potential to replicate, transition to the kidneys and the bloodstream. Each of these facets of host–pathogen interactions activates distinct paths of both innate and adaptive immunity, which may alter TBI outcome. LPS does elicit a strong pro-inflammatory immune reaction, but this occurs in the absence of an active infection. Therefore, UPEC is a preferential stimulus that reflects a true model of infection with multiple signaling pathways and defenses. This is critically important in defining the effects of post-injury immune insults in a translationally meaningful manner.
It is important to contextualize this work for translational relevance. These results should provide a broader understanding of the effects of UTI outside of the urinary tract system and a need to explore UTI’s effects on the brain and behavior. The experience of TBI, and perhaps other types of CNS injuries, could significantly influence the immune and behavioral response to UTI. This insight could reveal alternative intervention points to improve recovery in cases of comorbid UTI, with a focus on bladder–brain pathology. Additionally, many nosocomial UTIs are associated with indwelling catheters.65–67 There are established pre-clinical models of catheter-associated UTI, as well as recurrent UTI,68–71 and these could be explored in further studies. Finally, this work focuses on the role of post-TBI UTI in females. However, understanding the effects of UTI after TBI in males is of equal importance and should include an aging component.
In summary, UTI is one of the most common bacterial infections and frequently occurs in TBI survivors. Pre-clinical models offer a unique and valuable resource to define molecular pathways modulated by post-injury infection. Here, we established a combination model of TBI and UTI in female mice, demonstrating worsened cognitive recovery and elevated aspects of neuroinflammation at subacute post-injury timepoints. In addition to the points already discussed, future studies would benefit from increased sample size in the open-field test, as well as the consideration of estrous cycle as a biological variable. Together, these results show that the injured brain is vulnerable to the influence of peripheral infection. A better understanding of the neuroimmune and behavioral effects of post-injury bladder infection may uncover additional intervention points that improve long-term recovery.
Data Transparency Statement
A dose–response study was performed first to track the progression of the infection from bladder to kidney. Mice received either a moderate LFPI or a Sham injury. At 3 DPI, mice were inoculated transurethrally with either 1.0 × 106 CFU or 1.0 × 107 CFU of UTI89 UPEC. At the designated endpoints (4, 10, and 17 DPI), mice were euthanized by CO2 asphyxiation, kidneys and urinary bladders were dissected and cultured for E. coli presence. Animal numbers were n = 3 for the TBI groups and n = 2 for the Sham groups. A control naïve mouse was added to show that the vehicle and inoculation itself did not cause an infection. No mice were excluded from this study. Next, we examined the behavioral and immune response to 1.0 × 106 CFU of UTI89 E. coli following TBI because it remained localized to the bladder during the dose–response study. Separate cohorts of mice received either a moderate lateral LFPI or a sham injury. At 3 DPI, mice were inoculated transurethrally with either vehicle (0.01 M PBS) or 1.0 × 106 CFU of UTI89 UPEC, resulting in a 2 (Sham, TBI) × 2 (Vehicle, UTI) factorial design. Three mice were excluded from this cohort: two mice due to death from injury complications and one mouse due to an adverse reaction to inoculation. Y-maze was performed 6 DPI with an n = 65 (16–17 per experimental group). The three mice mentioned prior were also excluded from the Y-maze data as well. Investigators were blinded to both running and scoring these behavioral tests. At 6 DPI following Y-maze, a subset of animals (n = 6–8 per experimental group) was euthanized by CO2 asphyxiation. Blood and brain ipsilateral to the injury were collected to examine immune cells. Blood was centrifuged, and plasma was collected for analysis of plasma cytokines. Blood and urinary bladders were also plated on LB agar plates to assess the presence of E. coli. One mouse was also excluded from the brain and blood flow cytometry analysis due to a processing error but was included in righting time, bacteria plates, plasma cytokine, and behavioral testing analysis. The remaining mice (n = 9 per experimental group) were then euthanized by CO2 asphyxiation, and brains and urinary bladders were dissected and processed for IF histology 10 DPI. Due to unexpected tissue integrity complications, one cohort of tissue (both brain and urinary bladders) could not be used for IF histology, leaving n = 6 per experimental group for both brain and bladder IF results. Investigators were blinded to experimental groups when imaging tissues as well as calculating percent area. Brain and urinary bladder results did originate from the same mice throughout the experiment. All procedures, protocols, and materials are described in the “Materials and Methods” section of the article.
Authors’ Contributions
Z.Z.: Conceptualization, formal analysis, investigation, methodology, project administration, validation, visualization, writing–original draft, and writing–review and editing. C.L.: Investigation, methodology, validation, and writing–review and editing. R.B., M.T., and A.M.: Investigation, validation, and writing–review and editing. H.S.: Validation and writing–review and editing. I.C.: Conceptualization, funding acquisition, methodology, validation, and writing–review and editing. D.M.: Conceptualization, funding acquisition, methodology, validation, resources, and writing–review and editing. O.N.K.-C.: Conceptualization, funding acquisition, methodology, project administration, resources, supervision, validation, visualization, writing–original draft, writing–review and editing.
Footnotes
Acknowledgments
The authors greatly appreciate the Hultgren Laboratory for sharing the UPEC and offering technical guidance on UTI procedures. The authors would also like to thank the Glasper Laboratory for the use of the Nikon microscope, and Zachary Weisenseel and Christopher Cotter for technical assistance with imaging and quantification of IF. Brain region inserts created in BioRender. Zimomra, Z. (2026) https://BioRender.com/nrweubk (mPFC); ![]() (cortex and hippocampus near injury site).
(cortex and hippocampus near injury site).
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by pilot funding from the Chronic Brain Injury Program and the Institute for Behavioral Medicine Research at The Ohio State University.