
Abstract
Thermosetting blends of 4-aminophenoxyphthalonitrile (APN)/epoxy resin (ER) were investigated by differential scanning calorimetry (DSC), dynamic rheological analysis and thermogravimetric analysis (TGA). The self-promoted curing behaviors and processing properties of the APN/ER blends and prepolymers were studied by DSC and rheological analysis under dynamic and isothermal conditions. The APN/ER blends and prepolymers exhibited two-step polymerization reactions in the absence of any other curing additives. The APN/ER blends and prepolymers had large processing windows with low melt viscosity, and the size of the processing windows was dependent on the APN monomer concentration. The thermal behaviors of the APN/ER copolymers were studied by TGA. These cured copolymers exhibited decomposition temperatures over 367 oC in either flowing nitrogen or air, and high char yield at 800 oC of 68% under nitrogen atmosphere. Consequently, these studies revealed that the APN/ER system exhibits attractive self-promoted curing reaction, desirable processing feature, excellent thermal and thermal-oxidative stabilities, and high char yields. The APN/ER copolymer will be a good candidate as a matrix for high-performance polymeric materials.
1. Introduction
Blending technology which is well known for designing new polymeric materials has attracted considerable attention and has been widely explored. 1 This is because these new materials generally have a desirable combination of unusual properties such as high temperature resistance, high mechanical performance and good processability. Meanwhile, their properties can be tailored by varying the individual polymer components in blend composition. Recently, three different blending systems have been studied including thermoplastics/thermoplastics, thermoplastics/thermosets and thermosets/thermosets. It should be pointed out that some examples in the thermosetting systems have been investigated. 2
As high-temperature-resistant polymers, phthalonitriles have received increasing attention both from academia and industry. Owing to their rigid molecular structure, phthalonitriles exhibit high glass transition temperatures (T g), good dimensional stability, excellent moisture resistance and superior flame resistant properties, which makes them ideal candidates as materials for marine, aerospace and electronic uses. 3 –5 However, these phthalonitrile polymers have problems of poor processability due to high curing temperature, long curing time and narrow processing window (temperature between the melting point and the polymerization temperature). 6, 7 Therefore, there is growing interest in two research directions: (1) the utility of various curing additives is expected to initiate the curing process and shorten the curing time of the phthalonitriles; 8 –12 (2) phthalonitrile-based monomers/oligomers containing a variety of linkages are designed and synthesized to pursue low complex melt viscosity and broad processing window for advanced applications 13 –23 (Scheme 1). Thus, the addition of curing additives with their volatility at high temperatures has an influence on thermal properties of phthalonitrile-based compositions. 13 –21 Therefore, it is essential to improve the processability of the phthalonitriles without significantly sacrificing their other desirable properties; it is desirable to modify the structure of the phthalonitrile monomers to remove the utilization of curing additives; it is well known that a simple and effective method is to incorporate amino or hydroxyl groups into the reactive phthalonitrile units. Then, lots of amino-functional or hydroxy-functional phthalonitrile polymers have been proved to be cross-linked by self-promoted curing without the addition of any other curing additives. 8, 24
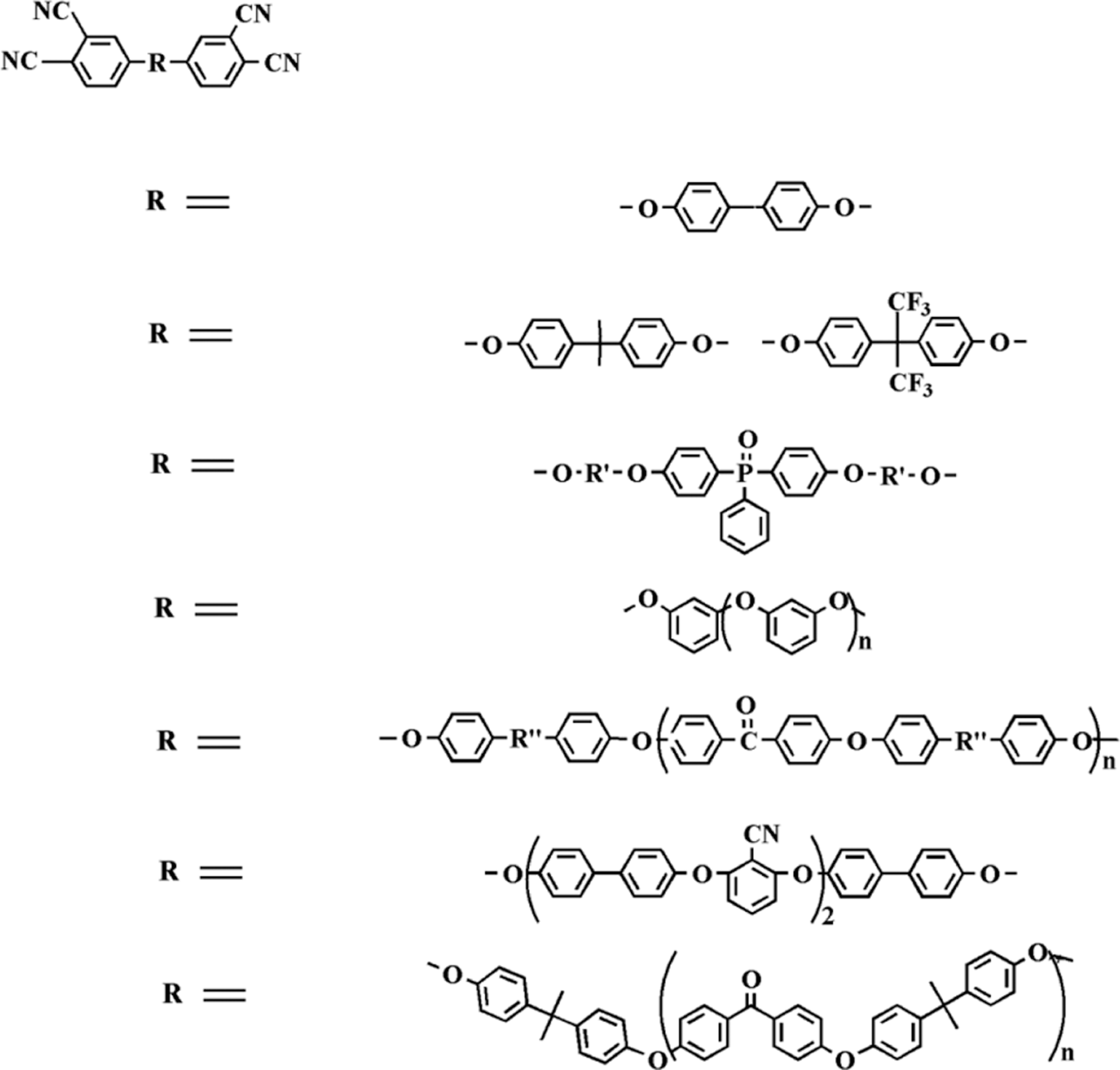
Phthalonitrile monomers.
In our lab, an aromatic amino group was incorporated into the phthalonitrile backbone with the reactive nitrile groups via a nucleophilic substitution reaction, and this resulted in the formation of an amino-containing phthalonitrile monomer. The monomer (4-aminophenoxyphthalonitrile, APN), which had low melt temperature (134 oC) and high curing temperature (295 oC), was successfully used as a kind of aromatic amine to achieve good processability of the APN/bisphthalonitrile blends. 12 Moreover, the blend composition maintained excellent thermal and thermal-oxidative stabilities. Therefore, the APN will be a good candidate for polymerization of epoxy resins (ERs) by blending technology.
ERs are well known for their outstanding properties such as moderate mechanical properties, good chemical resistance, great electrical properties and excellent processability. These characteristics make them very attractive to develop composites used in coatings 25 , adhesives 26 , electrical insulating materials 27 and aerospace composites 28 . However, they have low softening temperature, poor thermal and thermal-oxidative stabilities.
In this study, to expand the various applications of the APN with high performances in processing, thermal and flame-retardant properties, we will report a novel system. For this purpose, the APN and ERs were blended. The cure behaviors, rheological behaviors and thermal properties of as-prepared blend were systematically investigated. Then, there has been considerable interest in the APN with amino groups and phthalonitrile units, because it is not only able to catalyze the polymerization reaction of ERs, but also shows self-promoted curing behaviors to provide improved thermal and flame-retardant properties. These studies revealed that the APN/ER system exhibited attractive self-promoted curing polymerization, desirable processing features and excellent thermal stability.
2. Experimental details
2.1. Materials
APN was prepared according to the reference reported previously. 22 ER (diglycidyl ether of bisphenol A, DGEBA) with epoxide equivalent weight of 213–244 was kindly supplied by Blue Star New Chemical Material Co. Ltd. All starting materials were used without any further purification.
2.2. Preparation of APN/ER blends
A typical procedure for the preparation of the APN/ER blends was as followed: APN and the viscous liquid ERs were mixed using a stirrer at a stirring speed of 300 rpm to form a homogeneous mixture. After stirring at 50–70 oC for 30–45 min, the mixture was cooled to room temperature for differential scanning calorimetry (DSC) and viscosity studies. Then, the same mixture was treated by heating in an oven at 135 oC for 1 h, 2 h and 4 h, respectively, under a nitrogen atmosphere. The APN/ER blends were formulated with 25:75, 50:50 and 75:25, in which the numbers represented the weight percentages of APN and ERs, respectively.
2.3. Preparation of APN/ER prepolymers and copolymers
The APN/ER prepolymers were prepared under normal nitrogen atmosphere by adding the APN monomer into the liquid ER at 120–140 oC, achieving a dark melt. After stirring vigorously for about 20–25 min, the homogeneous melt was cooled rapidly to room temperature. The prepolymers were pulverized for viscosity studies. The same melt was poured into a pre-heated polytetrafluoroethylene mold with cavity dimensions 65 mm × 15 mm × 5 mm. The samples were thermally cured in a nitrogen-circulation oven at 200 oC for 1 h, 240 oC for 3 h, 260 oC for 2 h and 280 oC for 1 h. Then, the obtained cured APN/ER copolymers were pulverized for thermogravimetric analysis (TGA).
2.4. Characterizations
Fourier-transform infrared (FTIR) spectra were recorded with a Shimadzu FTIR8400 S Fourier Transform Infrared spectrometer in KBr pellets between 4000 and 400 cm-1 in air. Dynamic DSC analysis was employed to study the cure process of the blends using a TA Instruments Modulated DSC-Q100 at a heating rate of 10 oC min−1 and a nitrogen flow rate of 50 ml min−1. Viscosity studies of the blends and prepolymers were conducted using TA Instruments Rheometer AR-G2 with a heating rate of 3 oC min−1, at a frequency of 1 Hz. The samples (0.5–1 g) were melted between 25 mm diameter parallel plates with an environmental testing chamber of the rheometer. TGA of cured copolymers was determined by TA Instruments Q50 with a heating rate of 20 oC min−1 under nitrogen or air atmosphere with a purge of 40 ml min−1.
3. Results and discussion
3.1. Self-promoted curing behaviors on APN/ER blends under dynamic conditions
The APN was synthesized according to the one-step reaction sequence, as depicted in Scheme 2. The amino group of 4-aminophenol was incorporated into the 4-nitrophthalonitrile backbone with the reactive nitrile groups via a nucleophilic substitution reaction and this resulted in formation of an amino-containing phthalonitrile monomer (Scheme 2). Its molecular structure was characterized by FTIR spectroscopes. FTIR (KBr, cm−1): 3443 cm−1 (–NH2), 2230 cm−1 (–CN), 1248 cm−1 (stretch, C–O–C), 1477 cm−1 (1, 2 and 4 substitution of benzene ring), 849 cm−1 (1, 3 substitution of benzene ring). Owing to its reactive amino groups and nitrile groups, there has been increasing interest in the synthesized phthalonitrile. It is not only able to catalyze the ring-opening polymerization of ERs, but also shows self-promoted curing behaviors in the absence of any other curing additives.
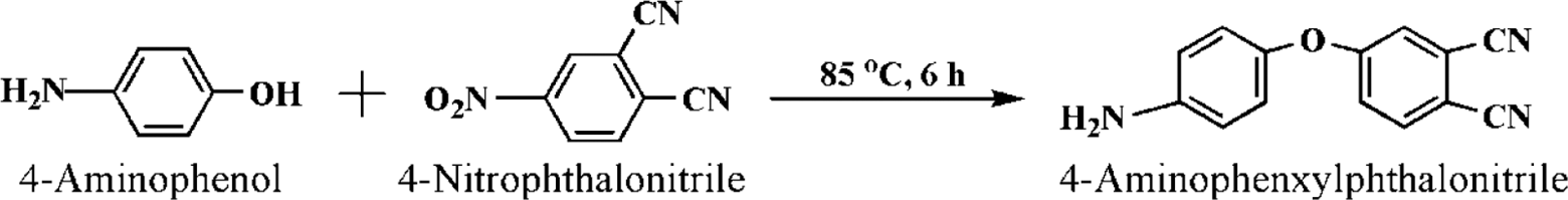
Preparation of the APN monomer.
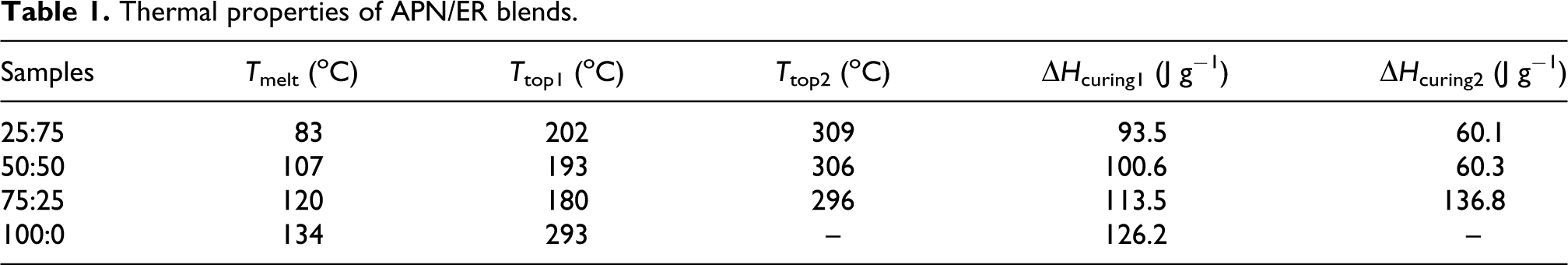
The self-promoted curing behaviors of the APN/ER blends were studied by DSC analysis. DSC curves from 50 to 350 oC of the APN/ER blends prepared at room temperature are presented in Figure 1, and DSC results are summarized in Table 1. The curves each exhibited an endothermic transition observed at 83–120 oC (T melt), which corresponded to the melting of the blends. The peak melting temperatures of the blends were dependent on the composition of the blend. ERs are a mixture of linear oligomers in the amorphous phase without a single melting peak. The APN monomer exhibited a single characteristic endothermic peak (134 oC), which is attributed to the melting transition. The degree of crystallinity for the APN monomer was determined from the melt enthalpy (78.6 J/g). Then, it was evident from the curves that the APN/ER blends exhibited one melting peak due to the existence of one predominant component, and the peak shifted to higher temperature with an increasing APN content. As shown in Figure 1, the APN monomer had one exothermic transition, which peaked at 293 oC. The exothermic transition corresponded to the polymerization reaction of the APN monomer. This is related to the cyclotrimerization of the phthalonitrile via its amino groups and nitrile groups. 3, 12 The curves of the blends exhibited two exothermic transitions that peaked in the range of 180–202 oC (T top1) and 296–309 oC (T top2). The first exothermic transition was due to the ring-open polymerization reaction of ERs via the amino groups of the APN monomer. A detailed reaction mechanism is shown in Scheme 3. Meanwhile, the exotherm peak temperature shifted with gradual decrease as the APN content of the blend increased. Presumably, this is because the increase of amino group concentration enhanced the supply ability of the active hydrogen. As the active hydrogen content of the blend increased, the start temperature of ring-opening polymerization of ERs decreased. Then, the latter exotherm was ascribed to the polymerization reaction of nitrile groups of the APN monomer. Similarly, the polymerization could be easily controlled by varying the APN monomer concentration. DSC results showed that the exotherm peak temperatures are a function of the APN content. In addition, the large processing window (>100 oC) for the APN/ER blends was observed, implying that the blends could flow at relatively low temperature and react at high temperature.
Thermal properties of APN/ER blends.
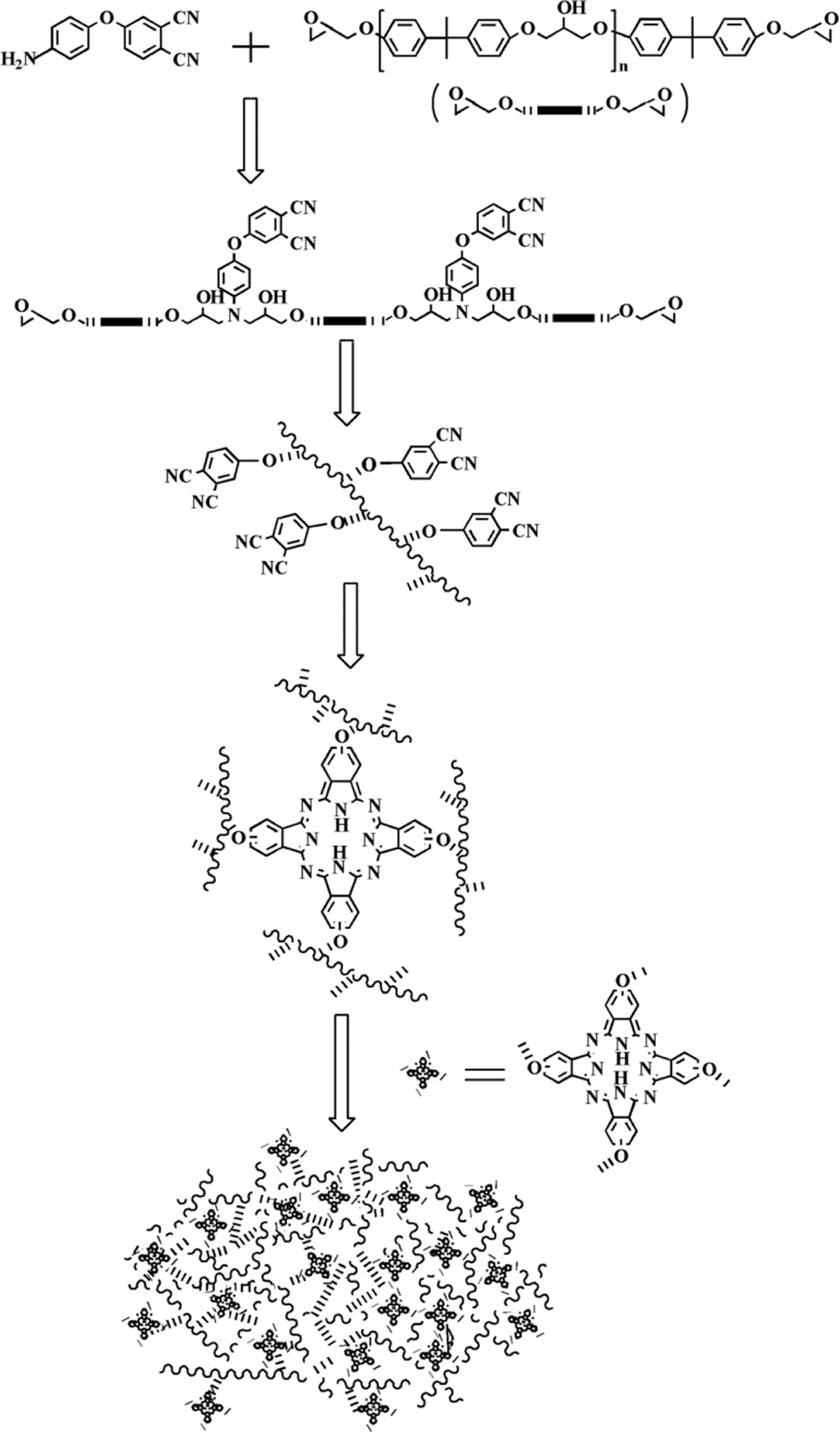
Preparation of the APN/ER copolymer.
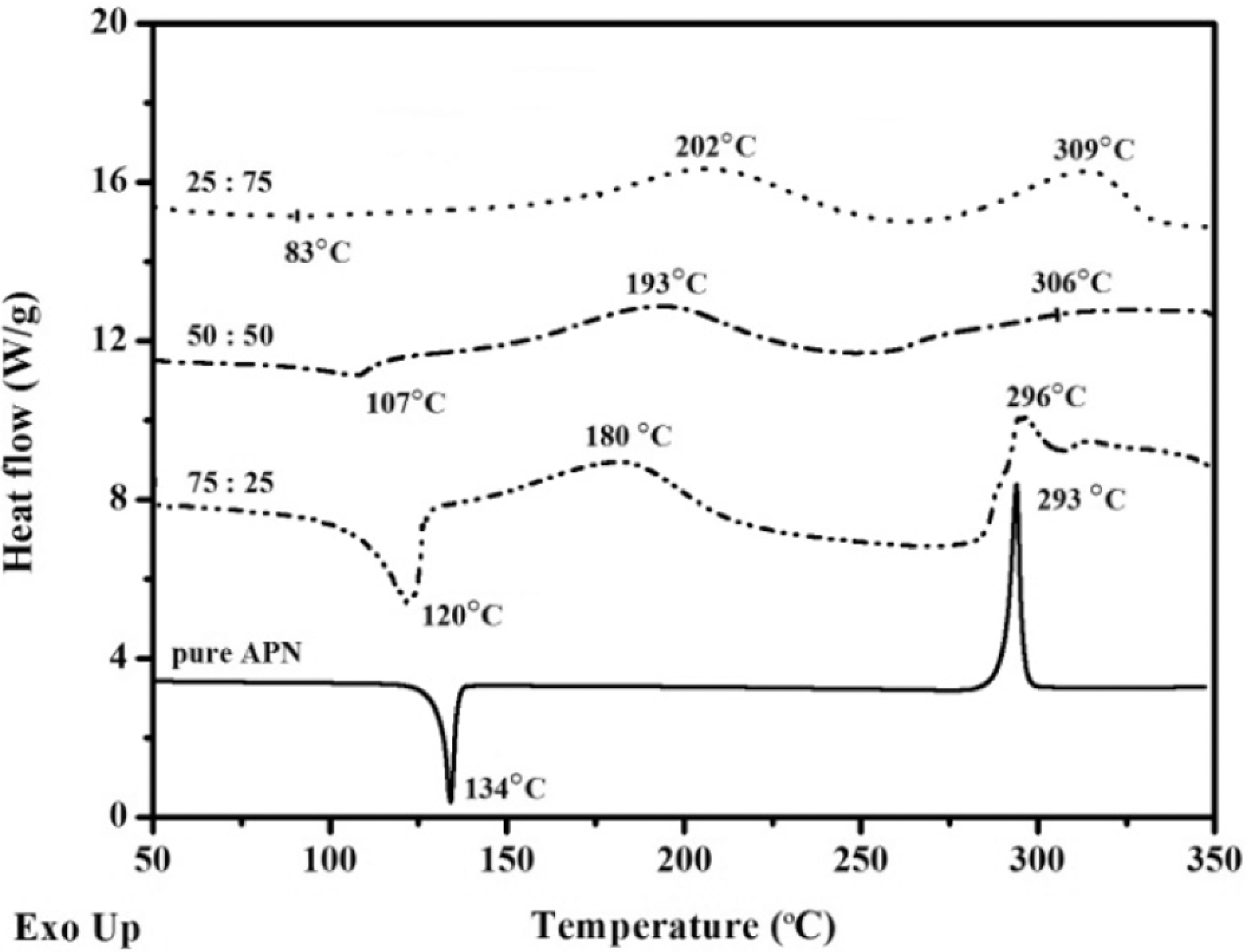
DSC curves of the APN/ER blends.
The rheological behavior, a key factor in predicting processability, was performed on viscosity changes accompanying the self-promoted curing reaction of the APN/ER blends. In Figure 2, the complex viscosity of the APN/ER blends was determined as a function of temperature from 100 to 350 oC. In each case, a rapid decrease was observed in viscosity on melting at 120 oC and a minimum melt viscosity of 0.01–1 Pa·s was observed for the blend. Above 150 oC, a melt viscosity increase for the APN/ER (25:75) blend was shown, which indicated that the ring-opening polymerization of epoxy resins was progressing. Up to 200 oC, this is the second time that the APN/ER (25:75) blend exhibited abrupt viscosity increase. The rapid increase of viscosity implied that the polymerization of the APN monomer was occurring. In contrast, the APN/ER (50:50 and 75:25) blends displayed a rapid increase in viscosity on melting at 250 and 285 oC, respectively. Therefore, the results revealed that the melting process of the blends was divided into two steps, and the polymerization reaction of the APN/ER blend was a two-step reaction. In addition, the rate of the copolymerization of the APN and epoxy resins could be easily controlled by varying the temperature. Moreover, the low melt viscosity of the blends exhibited their good melt stability at high temperatures, which was evidence of their good processability in absence of any other curing additives. These phthalonitrile–amine compositions were also reported to have good processing features. The aromatic curing additives concentration and variety could be used to control the rate of the polymerization and the processing window. 12, 20 –23
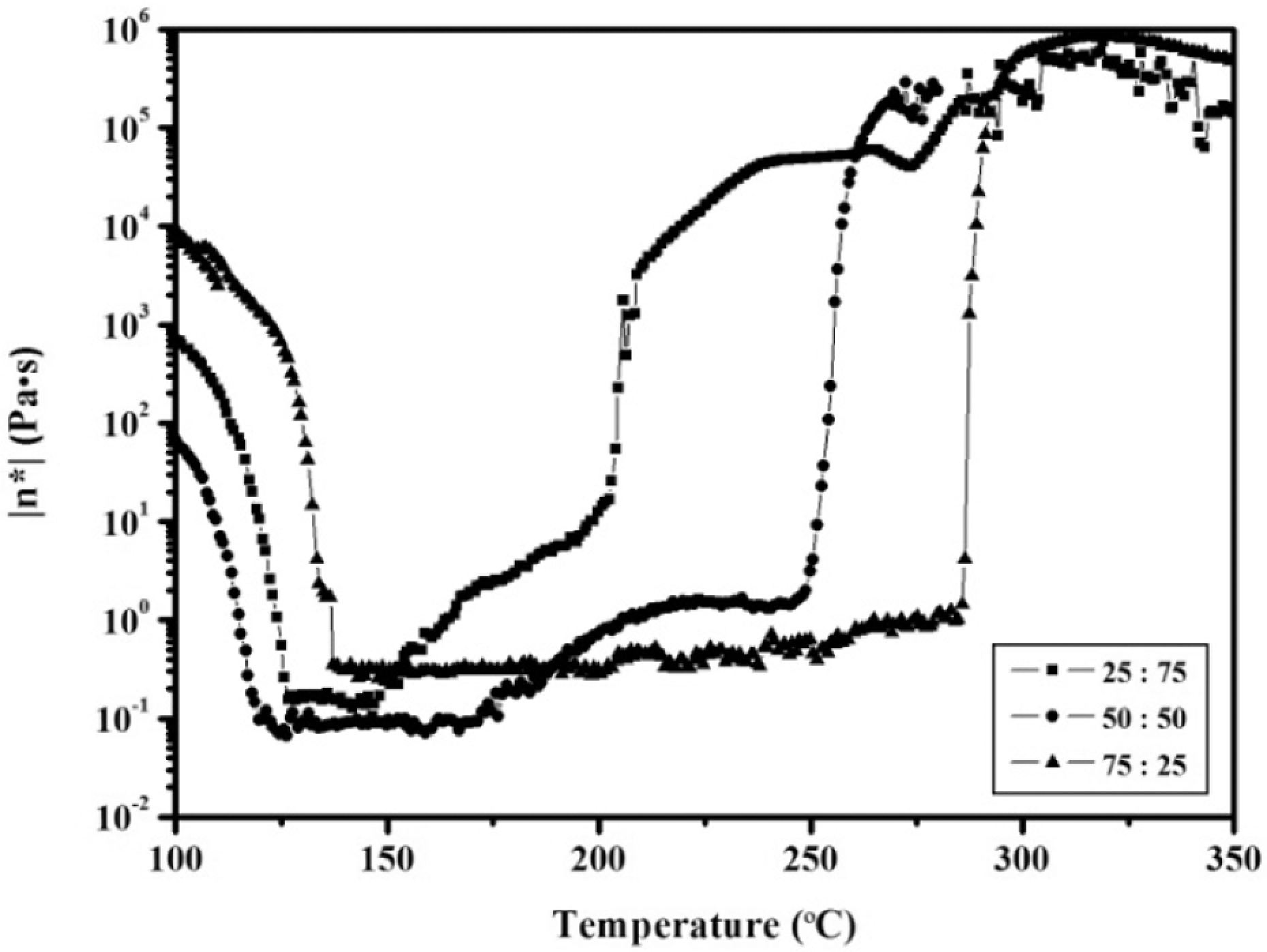
Complex viscosity (η*) as a function of temperature for the APN/ER blends.
To determine the initial temperature for polymer processing, complex viscosity changes of the APN/ER (50:50) prepolymer were noted as a function of time at several different temperatures. The curves shown in Figure 3 indicated that the higher the temperature, the faster the rate of viscosity increased. As expected, the result demonstrated that high temperature had a great tendency to accelerate the polymerization reaction of the APN/ER prepolymers. For comparison, isothermal viscosity measurements were recorded on the APN/ER prepolymers at 200 oC as a function of time. As depicted in Figure 4, a complex melt viscosity of around 0.1 Pa·s was observed for all of the prepolymers at 200 oC, initially. Then all of the prepolymers exhibited a dramatic increase viscosity when held at 200 oC. Obviously, the complex viscosity of the APN/ER (25:75, 50:50 and 75:25) blends increased to 500 Pa·s in about 18, 4 and 27 min, respectively. Therefore, these data revealed that the influence of the APN content on the rate of the copolymerization of the blend is evident and the blend has the desirable processing temperature and gelation time. In addition, the complex viscosity changes accompanying the self-promoted curing reaction of the prepolymer were monitored by viscosity studies. The time sweep curves of the APN/EP (50:50) prepolymer at 200 oC are depicted in Figure 5. As can be seen, the storage modulus (G′) and the loss modulus (G′) of the APN/ER (50:50) prepolymer increased as time increased. The G′ and G′ crossover point corresponding to the gelation time (gel point 29 ) was observed at 12 min. These results are consistent with the fact that the APN/EP (50:50) prepolymer had been transferred from a viscosity flow state to a solid state. 20 Meanwhile, the gel point corresponds to the start of cross-linking network formation, which is consistent with the results obtained in viscosity studies that the viscosity of the prepolymer increased abruptly as gelation occurred. In addition, the tan (delta) curve exhibited a sharp peak at 10 min, which suggested that the prepolymer can polymerized without any other curing additives under certain curing temperature. Based on these rheological results, the prepolymers exhibited the desirable polymerization rate and a long processing window, which are important to their applications in resin transfer molding or resin infusion processes.
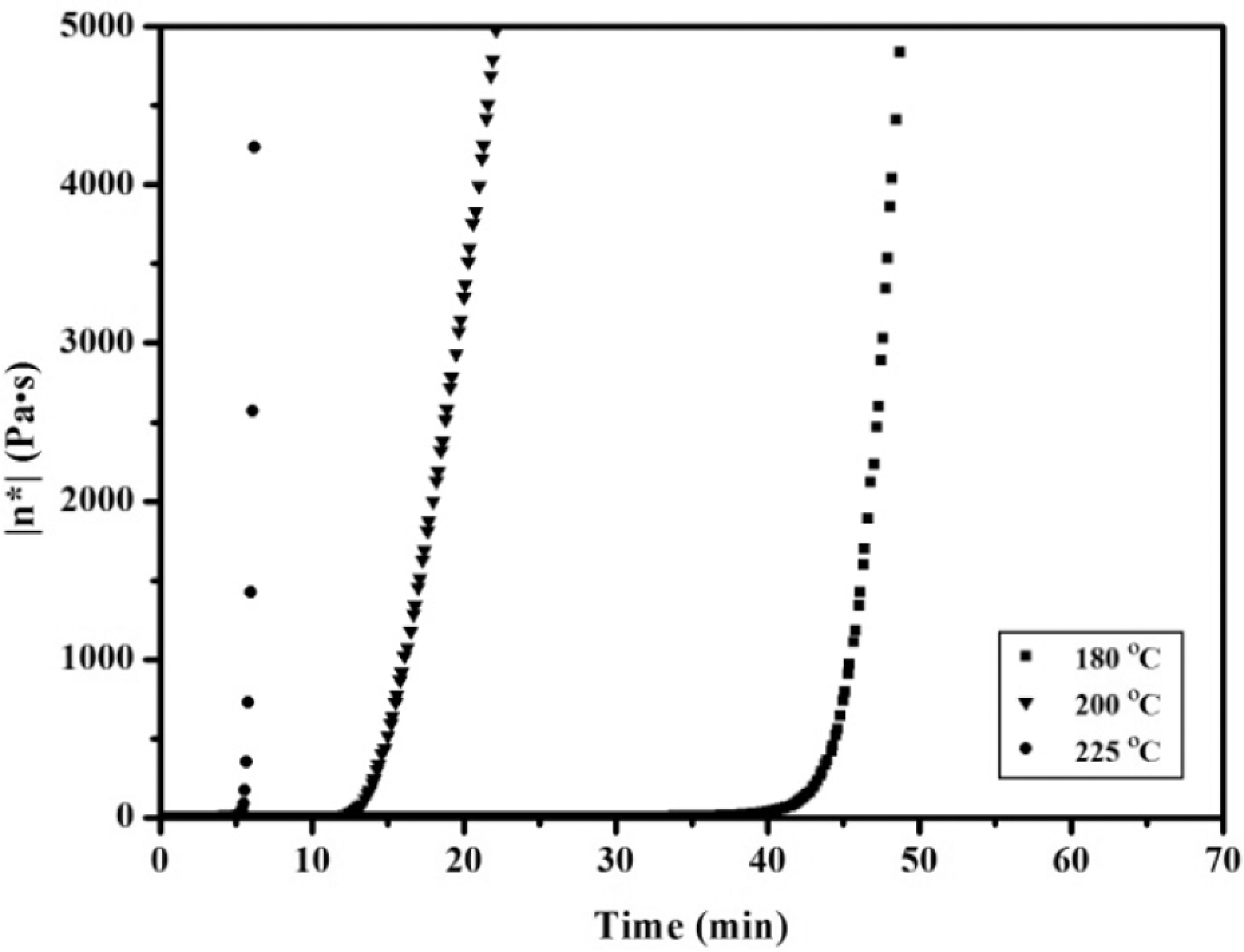
Complex viscosity (η*) as a function of time at various temperatures for the 50:50 APN/ER prepolymer.
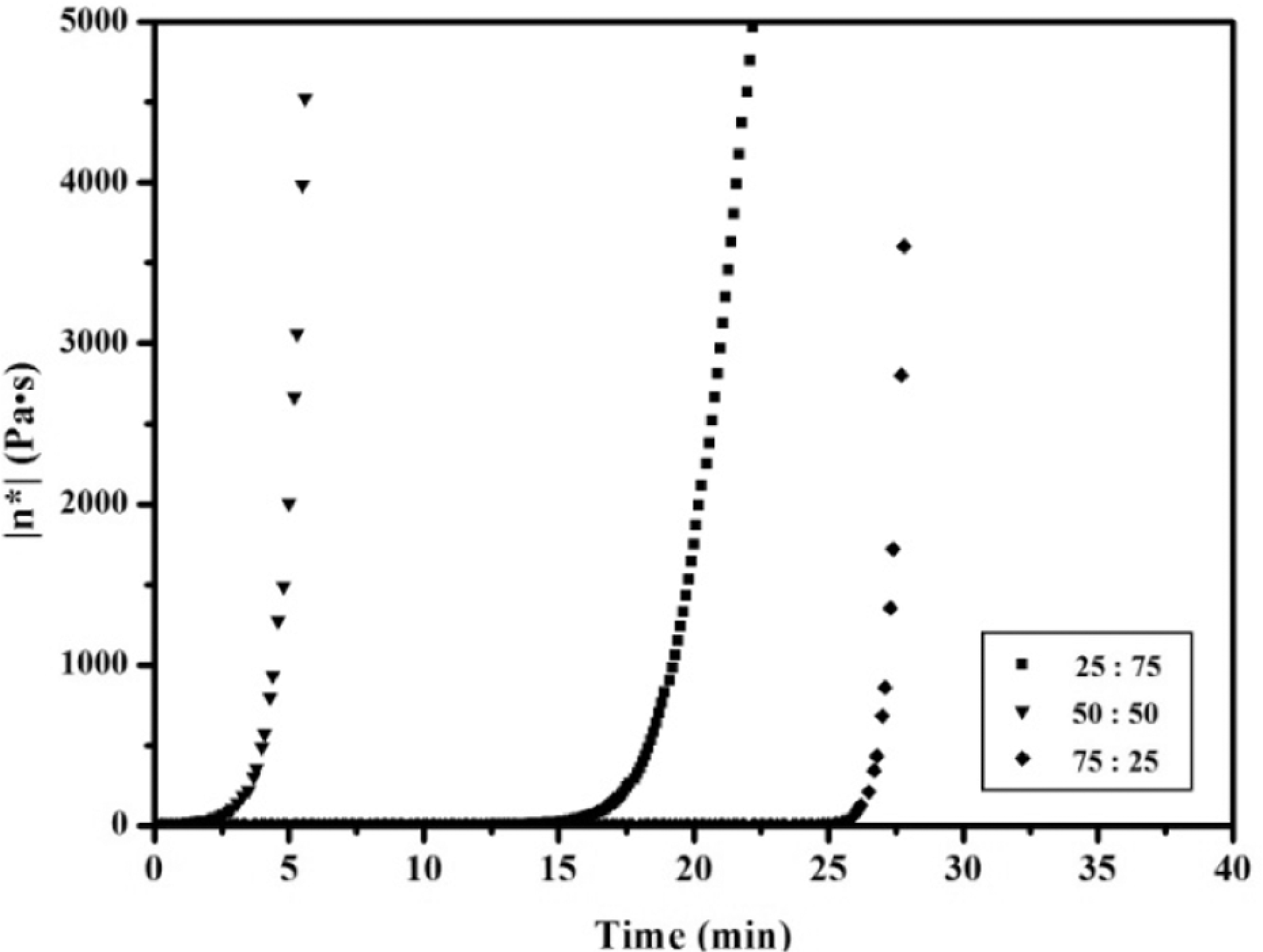
Complex viscosity (η*) as a function of time at 200 oC for the APN/ER prepolymers.
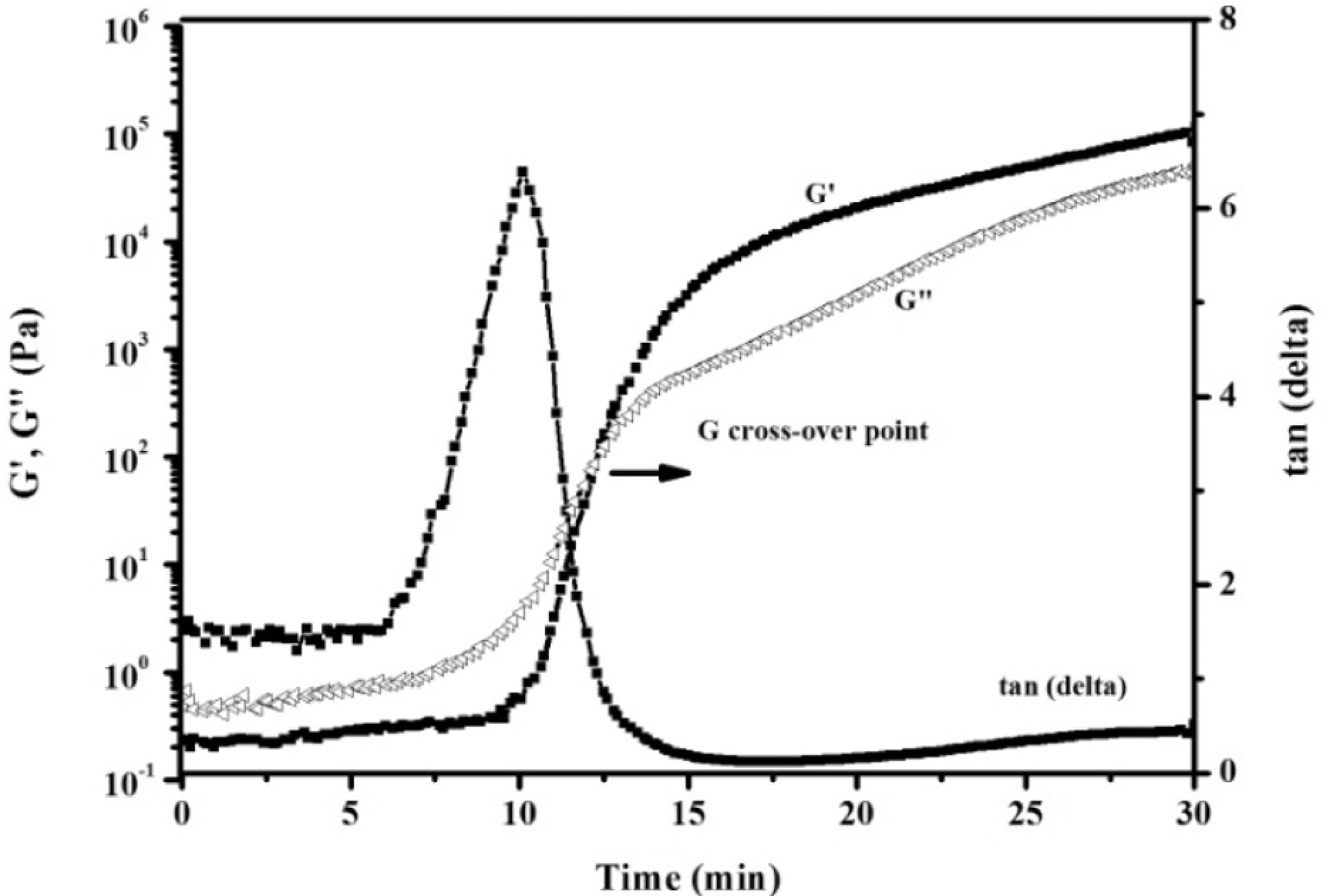
Time sweep curves of the 50:50 APN/ER prepolymer at 200 oC.
3.2. Self-promoted curing behaviors on APN/ER blends under isothermal conditions
Model curing studies were generally conducted to characterize the curing behaviors by differential analysis techniques. The dynamic and isothermal mode has been extensively used in DSC. The self-promoted curing behaviors of the APN/ER blends were evaluated under isothermal condition. Based on the dynamic curing data depicted in Figure 1, the temperature of the isothermal self-promoted curing measurements was evaluated as 135 oC, because the APN had a melting point of 134 oC, and the APN and ERs copolymerization onset temperature was about 140 oC.
The APN/EP (50:50) blend was treated at 135 oC for different times. As noted in Figure 6, no crystallization peaks for the samples were apparent, which indicated that the treated blends had been in the amorphous phase. For the APN/EP (50:50) blend at 135 oC for 1, 2 and 4 h, the peak of the step transition occurred at 65, 68 and 74 oC, respectively. It revealed that the treated blends possessed T g, and the T g values increased as the treatment time increased. For comparison, the appearance of the additional T g (at 207 oC) for the APN/EP (50:50) blend heated at 135 oC for 1 h indicated that the ring-opening polymerization reaction of ERs progressed with the start of copolymer network formation. Within the blends, the reactivity attributed to the melting APN seemed to depend on the treatment time. In addition, the treated blend (Figure 6: curve 135 oC for 1 h) exhibited a small exotherm centered at 275 oC due to the initial reaction of the APN monomer. These exothermic transitions for the treated blends were centered at 349, 353 and 355 oC, respectively. The transitions correspond to the thermal decomposition of the treated APN/ER blends and/or excess ER. The results, shown in Table 2, provided evidence that the cure reaction of ERs and the APN was processing very slowly at 135 oC. However, the treated blends still exhibited relatively low decomposition temperatures and char yields. In Figure 7, the decomposition temperatures (T 5%) were in the range of 307–329 oC and char yields were in the range of 27–37%. The results indicated that the heat-treated blends exhibited low thermal stability. Thus, the polymerization of the APN via its nitrile groups was not happening appreciably in the treated blends, because the good thermal stability of the phthalonitriles was attributed to the high aromatic nature of the system and the cross-linking density. Then, FTIR spectroscopy was used to offer some insight into the self-promoted curing reaction of the APN/ER blends after heating at 135 oC for 1, 2 and 4 h. The FTIR data revealed that the characteristic absorption peak of the nitrile band (2230 cm−1) decreased and the epoxy groups (920 and 859 cm−1) absorption disappeared. Meanwhile, weak peaks assigned to the triazine absorption (1520 cm−1) and phthalocyanine formation absorbance (1010 cm−1) were observed.
Thermal properties of the 50:50 APN/ER blends acquired after heating at 135 oC for various times.

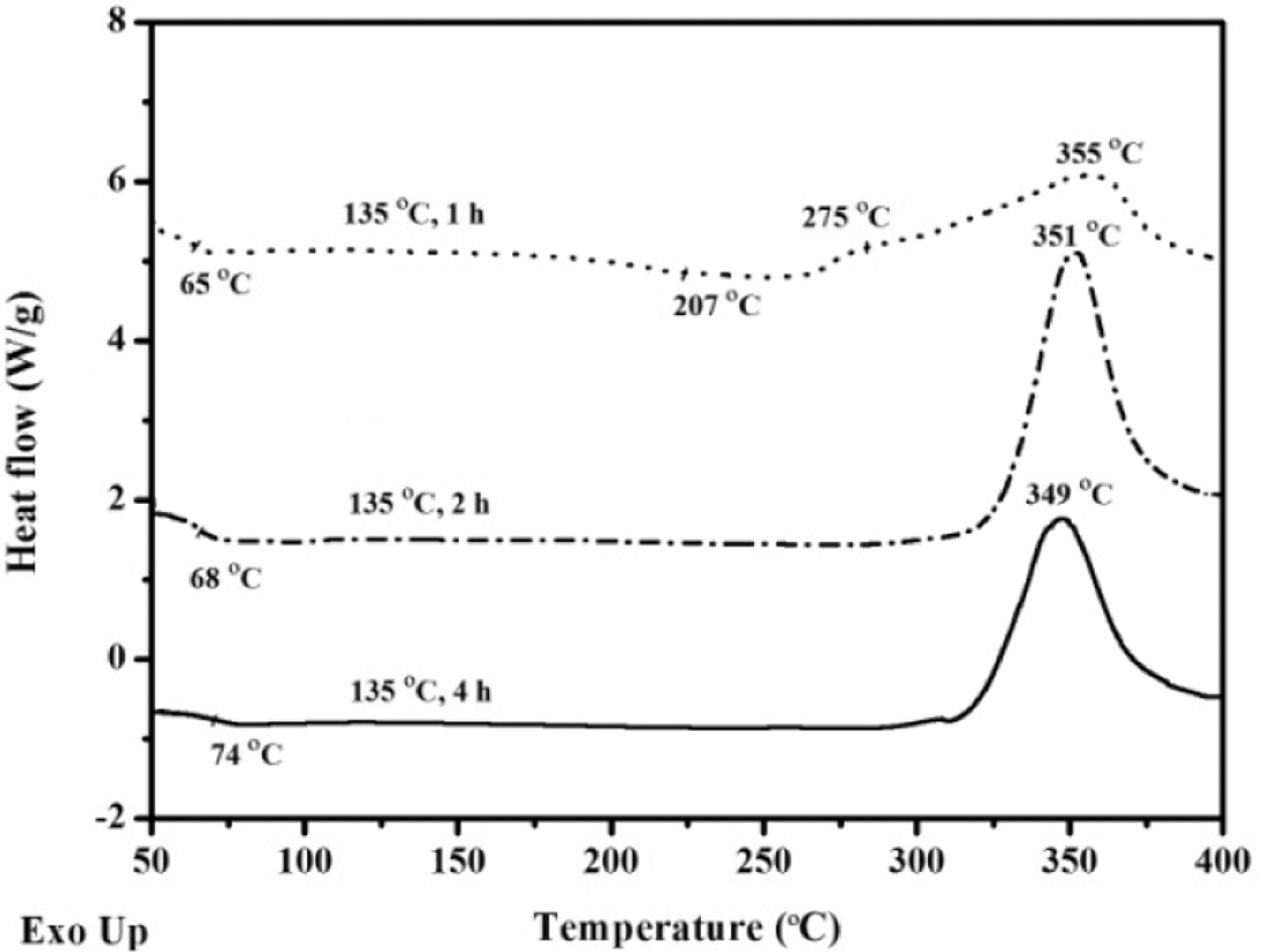
DSC curves of the 50:50 APN/ER blend acquired after heating at 135 oC for various times.
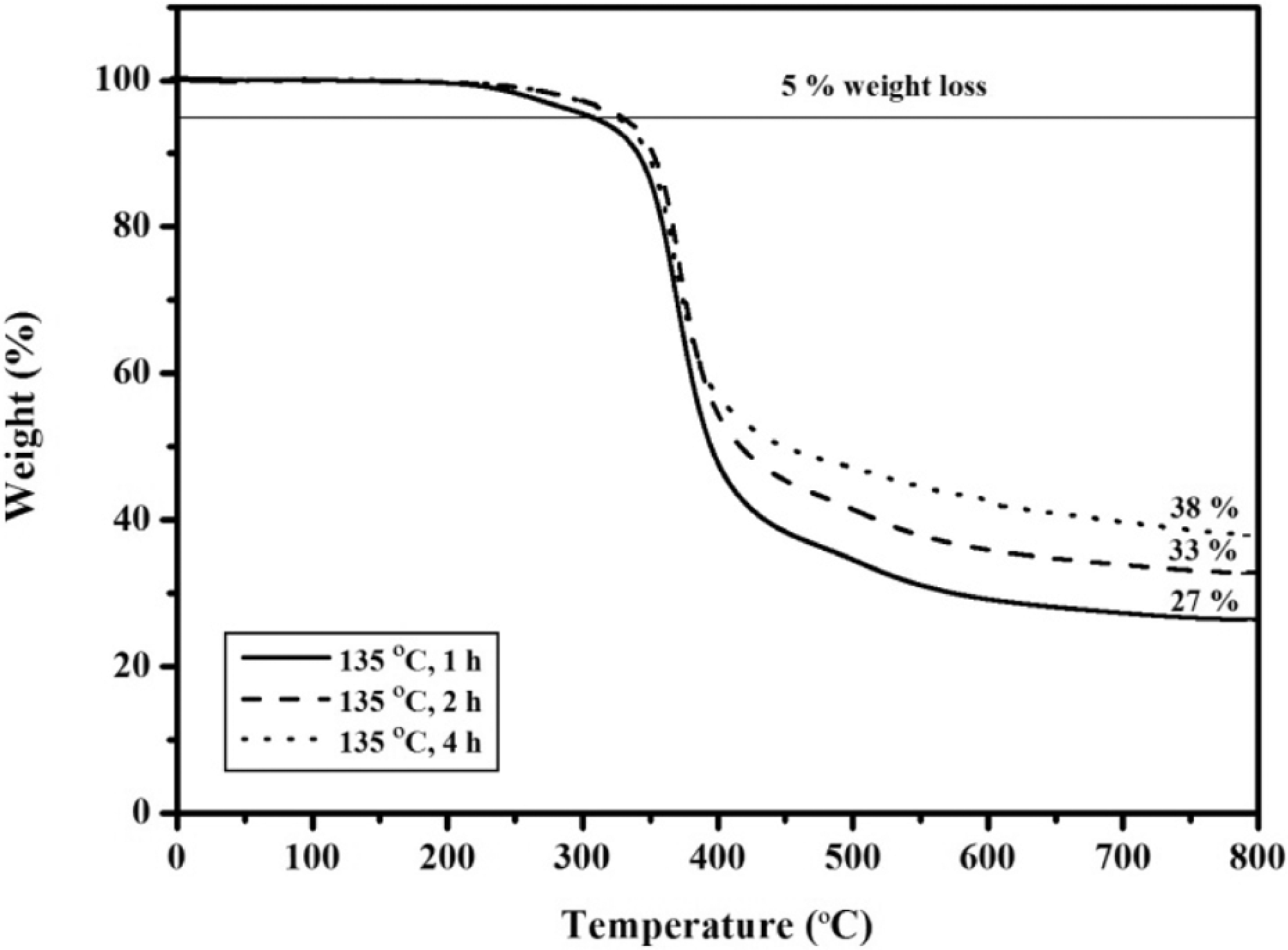
TGA curves for the 50:50 APN/ER blends acquired after heating at 135 oC for various times.
The cure of the APN/ER (50:50) blend acquired after heating at 135 oC for different times was also investigated by isothermal rheometric measurement at 200 oC. The curves, shown in Figure 8, indicated that the longer the heat-treated time, the faster the viscosity increase. This result demonstrated that the long heat-treated time had a great tendency to accelerate the polymerization reaction of the nitrile groups of the APN. Moreover, the time sweep curves of the treated blends at 200 oC are depicted in Figure 9. As can be seen, the delta curve of the treated blends exhibited one sharp peak at 43, 35 and 33 min, respectively. The peak shift to shorter time was evidence for that the reactivity of the treated blends increased as heat-treated time increased. These DSC and rheological data also indicated that the blends exhibited two-step reaction. In addition, polymerization rate control of the blends can advantageously affect their processing to a thermoset by moderate temperature and time.
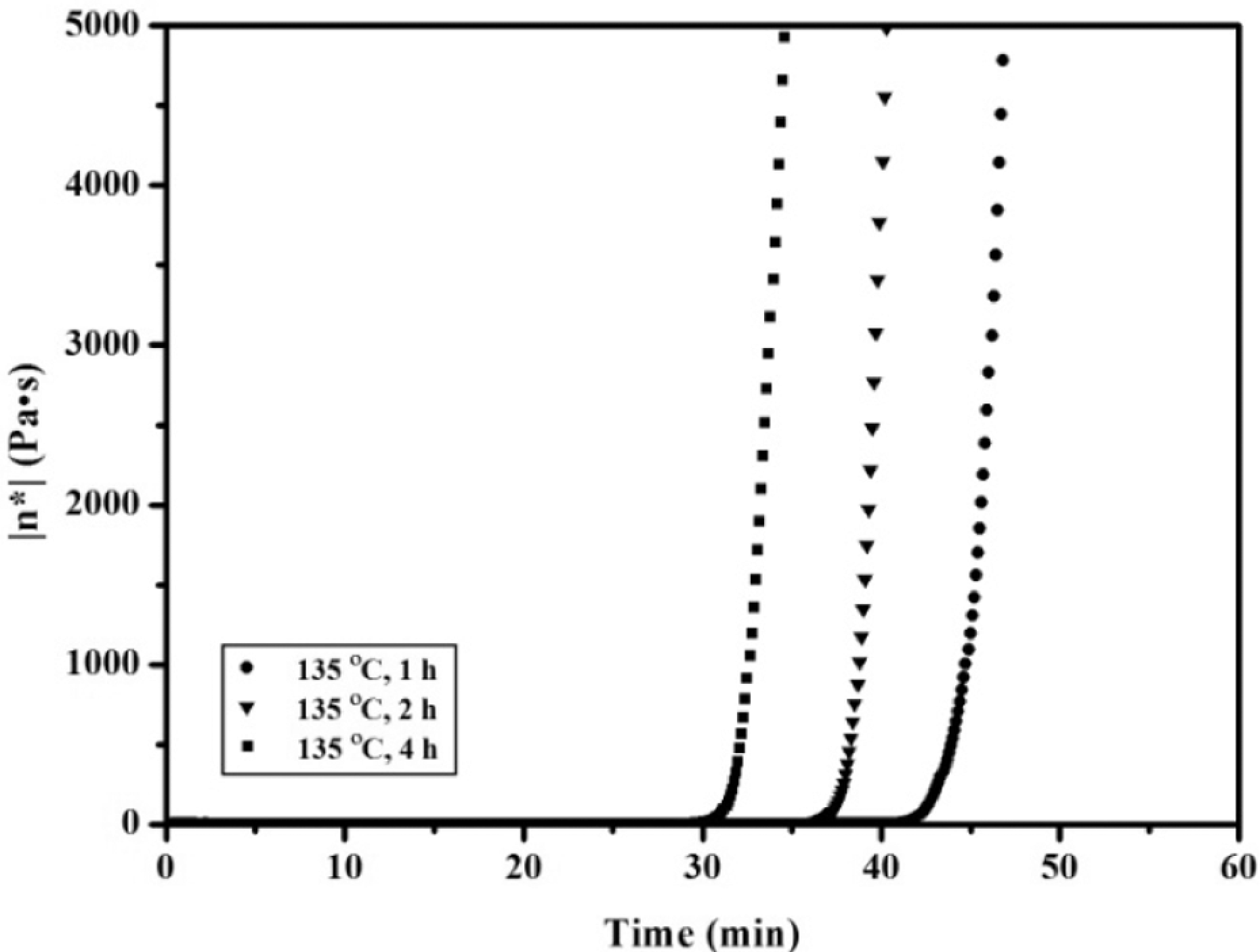
Complex viscosity (η*) as a function of time for the 50:50 APN/ER blend acquired after heating at different time.
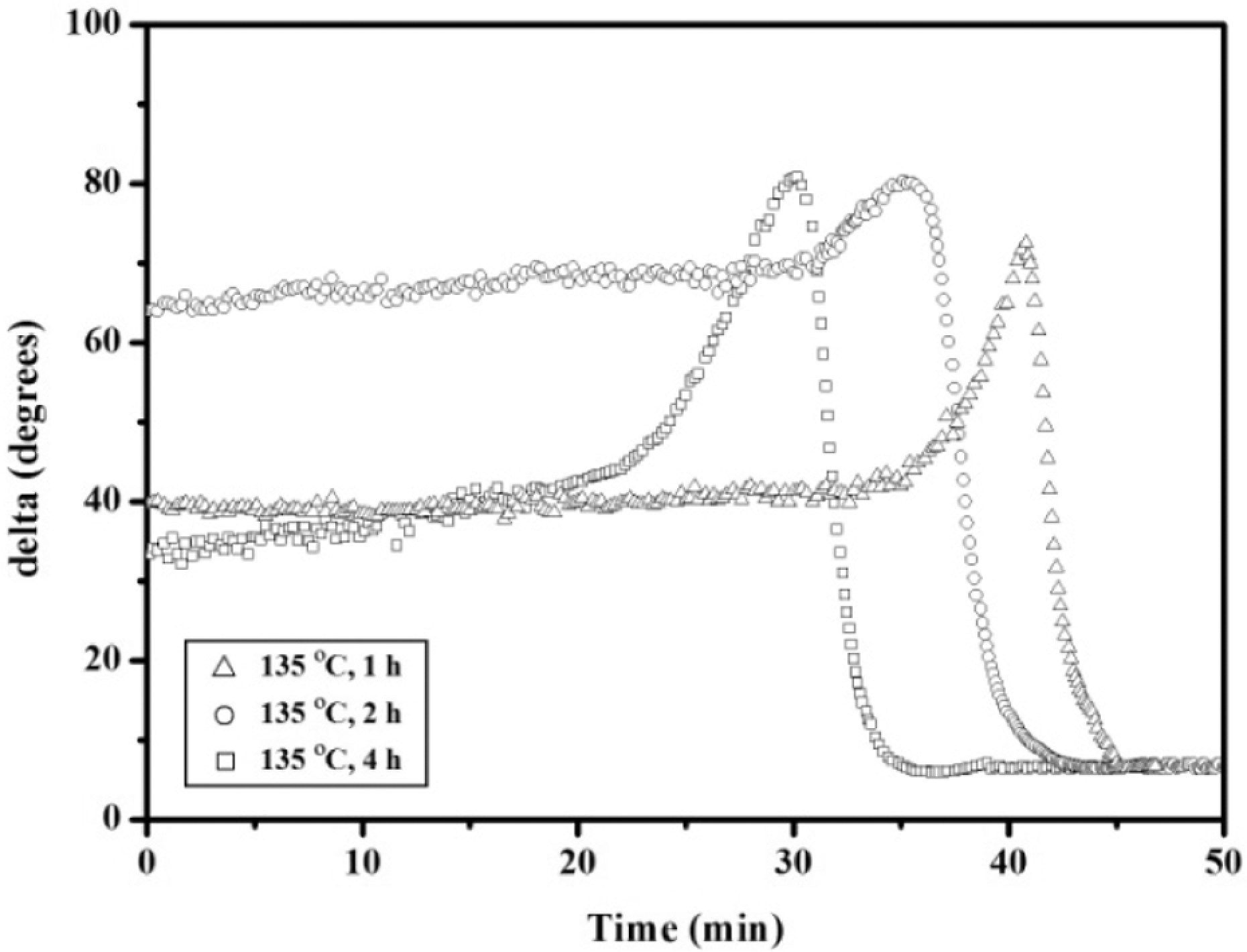
The delta curves for the 50:50 APN/ER blend acquired after heating at different time.
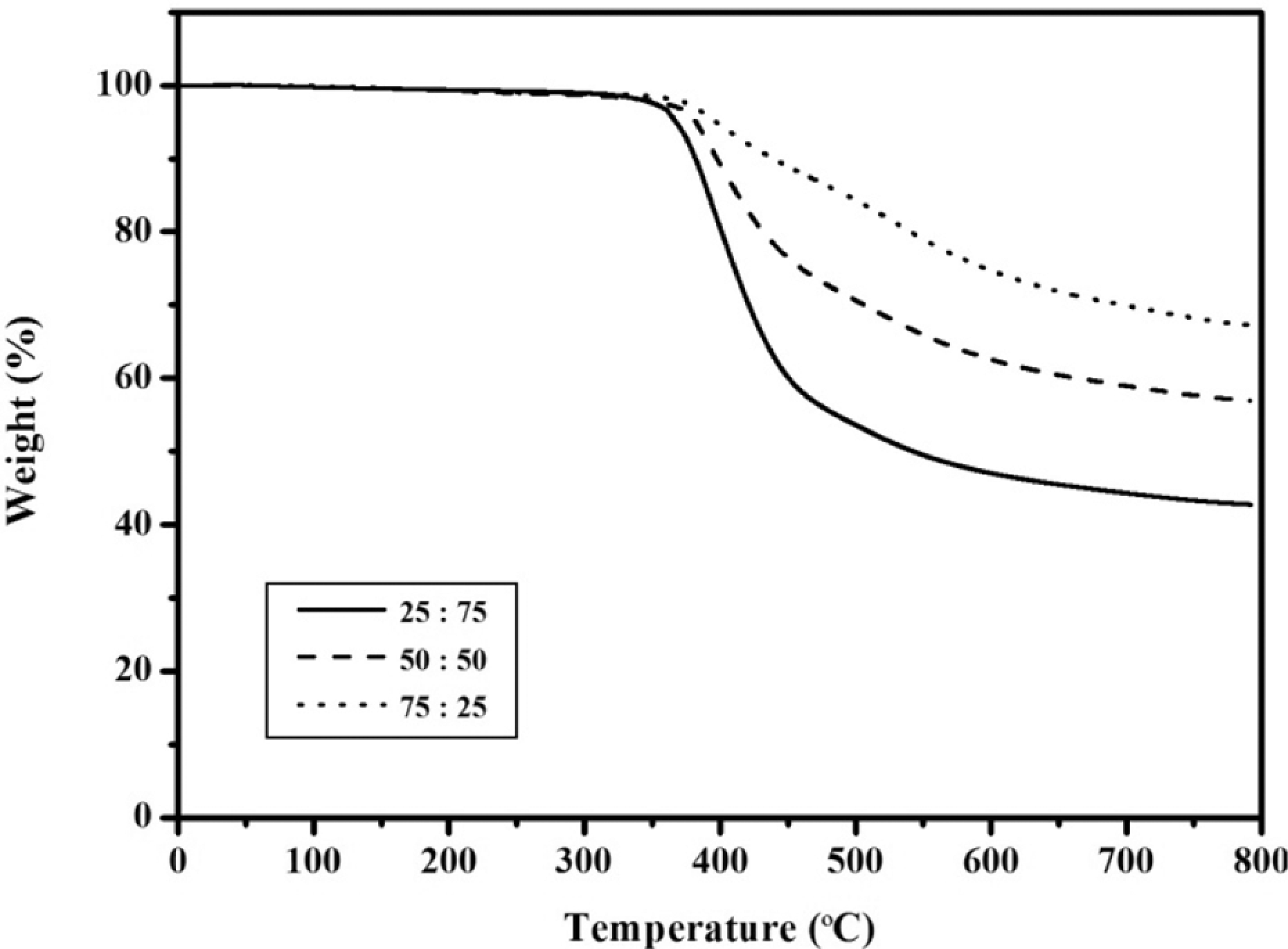
TGA curves of the APN/ER copolymers in nitrogen.
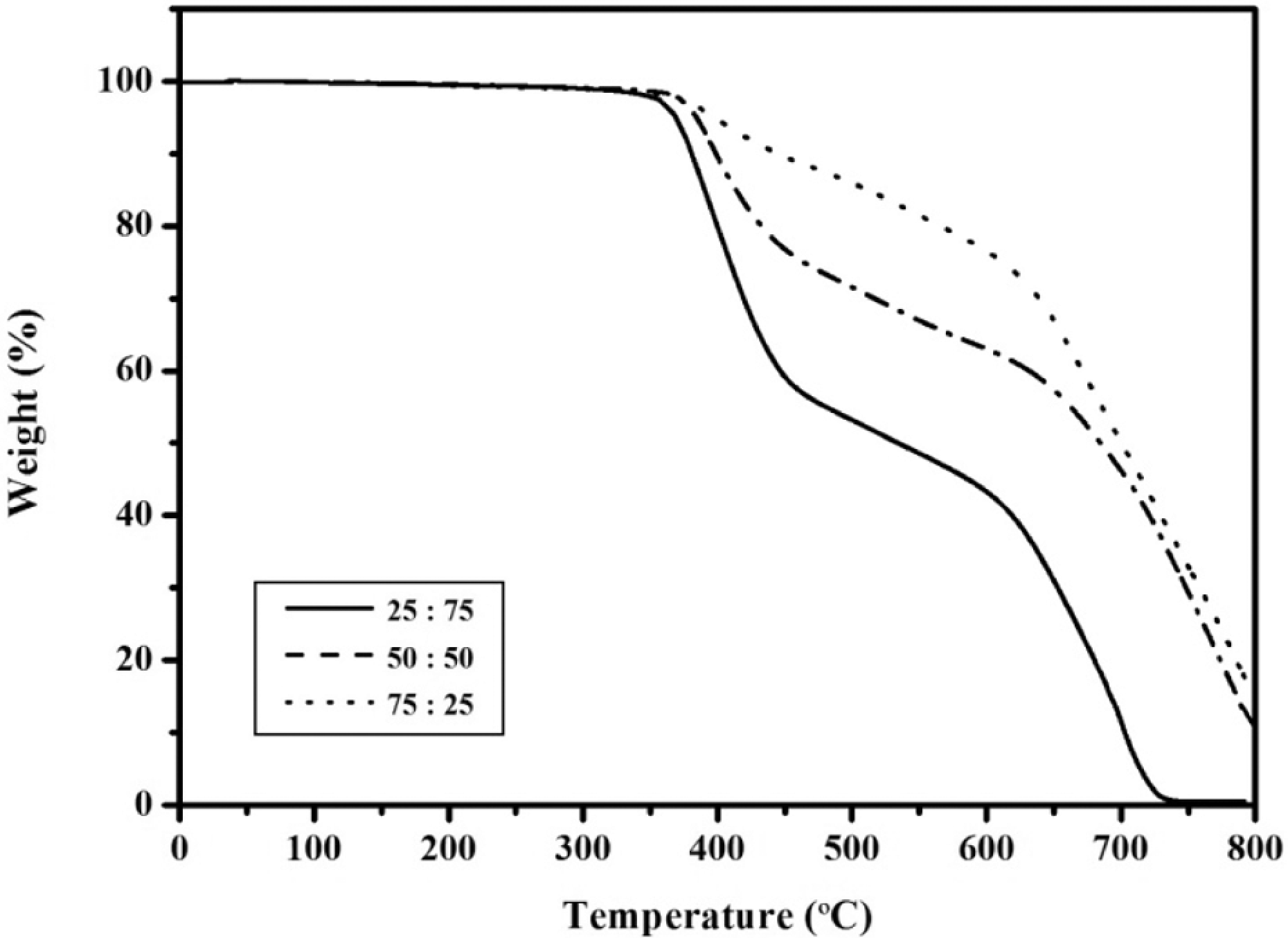
TGA curves of the APN/ER copolymers in air.
3.3. Thermal properties on APN/ER copolymers
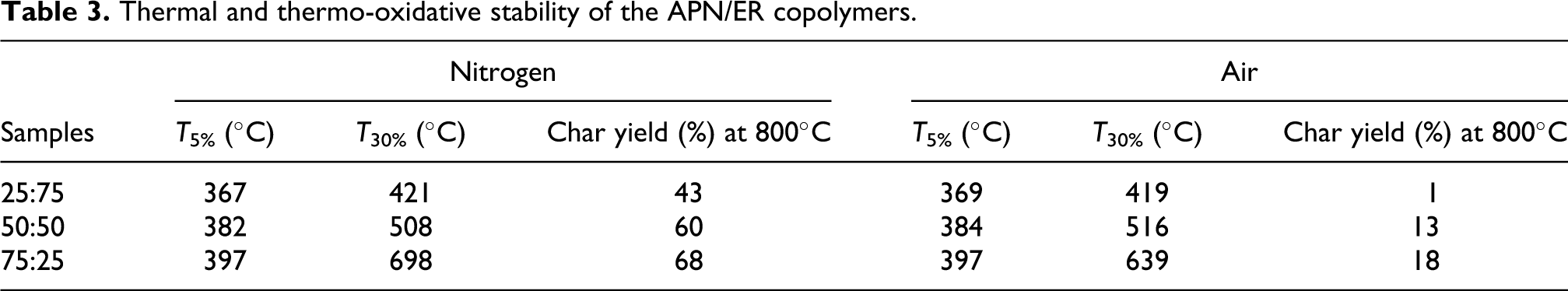
The thermal and thermo-oxidative stabilities of the APN/ER copolymers, without addition of any other curing additives, were determined by TGA analysis. In this study, the samples were cured by stepwise heating to 280 oC and holding at 280 oC for 1 h. The results of these studies were shown in Figures 10 and 11. The 5% and 30% weight loss temperatures (T 5% and T 30%) were measured, and the percentage of residue remaining (char yield %) after heating the samples to 800 oC in nitrogen and air atmosphere are listed in Table 3. The cured copolymers showed 5% weight loss temperatures in the range of 367-397 oC when heated in a nitrogen atmosphere. In air, these samples demonstrated T 5% in the range of 369–397 oC, respectively. According to these results, T 5% is somewhat higher in air atmosphere than that in nitrogen atmosphere, which may be ascribed to heterogeneity of the crosslink density. Moreover, these improvements in the thermal and thermo-oxidative stabilities were attributed to the increased cyano group concentration from the APN monomer. Obviously, the decomposition temperatures of the APN/ER copolymers were greatly improved compared with that of the neat ER. 30 The 43–68% and 1–18% char yields remained at 800 oC under nitrogen and air, respectively. Therefore, the APN/ER copolymers exhibited good thermal and thermal-oxidative stabilities, owing to the high aromatic nature of the system and the cross-linking density after curing. These data revealed that high thermal and thermal-oxidative stabilities of the APN/ER copolymers appeared to be very useful to the application.
Thermal and thermo-oxidative stability of the APN/ER copolymers.
4. Conclusions
The APN/ER blends, prepolymers and copolymers were prepared and characterized. The self-promoted curing behaviors of the APN/ER blends with varying APN contents were studied in the absence of any other curing additives. The APN/ER blends and prepolymers exhibited a two-step polymerization reaction determined by isothermal DSC and viscosity measurement. The APN/ER blends had large processing windows with low melt viscosity, and the size of the processing windows was related to the APN monomer concentration. The thermal properties of the APN/ER copolymers were also evaluated. TGA results showed that the copolymers exhibited excellent performance such as desirable processing features, high thermal stability, high thermal-oxidative stability and char yield. It is believed that the APN/ER copolymer can be used as a matrix for advanced composites with high performance.
Footnotes
Funding
The authors are grateful to the Major Science and Technology Project in Sichuan Province (2010 FZ 0117) and "863" National Major Program of High Technology (2012AA03A212) for the financial supports of this work.