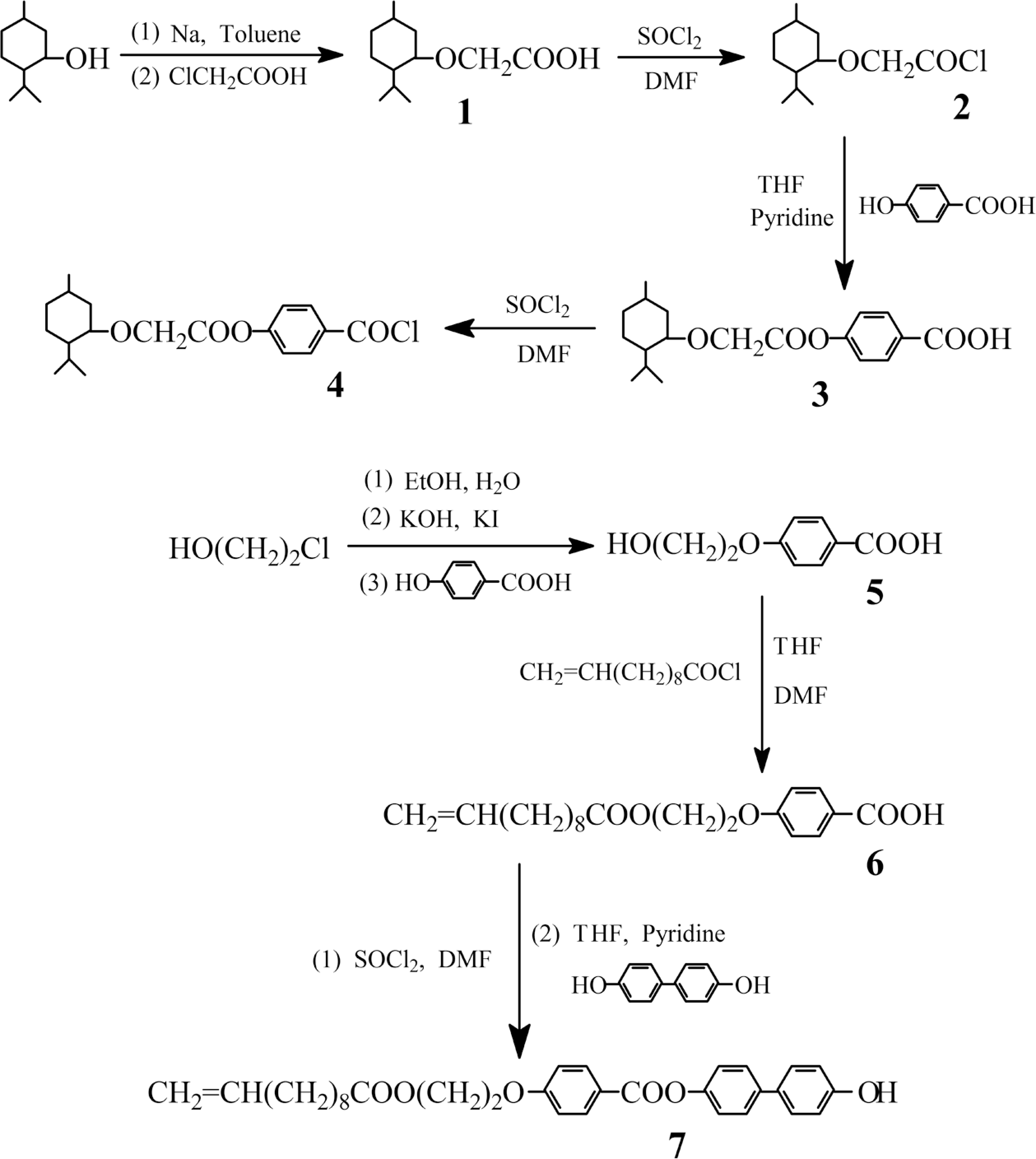
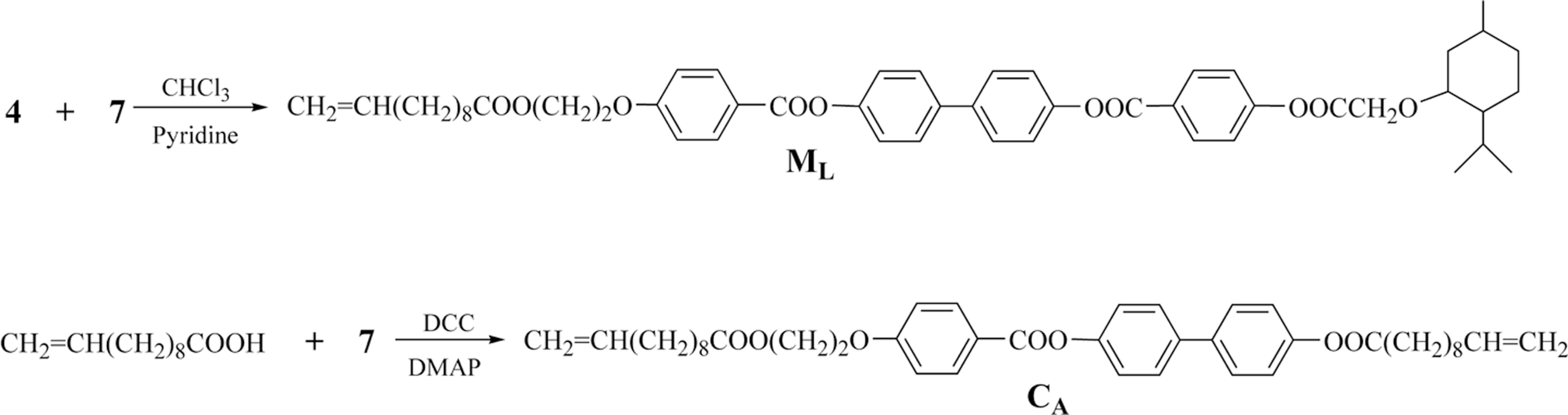
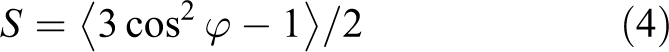The synthesis of new chiral monomer 4-(menthyloxyacetoxy- benzoyloxy)biphenyl-4′-(2-(undec-10-e noyloxy)ethoxy)benzoate (ML), crosslinking agent 4-(undec-10-enoyloxy)biphenyl-4′-(2-(undec-10-enoyloxy)ethoxy)benzoate (CA), and liquid crystal polymer networks (E1−E5) containing menthyl group is presented. Their chemical structures and phase behavior were characterized with Fourier transform infrared (FT-IR), proton nuclear magnetic resonance (1H-NMR), elemental analyses, polarizing optical microscopy, differential scanning calorimetry, thermogravimetric analysis (TGA), and X-ray diffraction. The selective reflection of light for ML was investigated with ultraviolet/visible/near infrared (UV/Visible/NIR). By inserting a flexible spacer between the mesogenic core and the terminal menthyl groups, MLcould form mesophase and show a chiral smectic C phase, cholesteric phase and cubic blue phase. CA displayed a smectic A phase and nematic phase. The polymer networks containing less than 12 mol% of the crosslinking units showed reversible cholesteric phase transition, wide mesophase temperature range, and excellent thermal stability. With increasing the content of crosslinking unit, the corresponding Tg increased, the Ti decreased, and the mesophase temperature range narrowed for E1−E5. TGA showed that the Td(5%) was greater than 330°C for E1−E5.
Today, liquid crystal (LC) materials are directed toward the development of multifunctional structure. One example of such supermolecular systems is liquid crystal networks (LCNs). Over the last decade, research on the LCNs has expanded rapidly.1–12 According to the crosslink density, two classes of LCNs emerge: highly crosslinked liquid crystal thermosets and lightly crosslinked liquid crystal elastomers (LCEs). Recently LCEs have been paid increasing attention because they give rise to new macroscopic features by combining the mechanical properties of polymer networks with the anisotropic structure of the LC phase, which make them candidates for several applications, such as flexible displays, electromechanical actuators, and artificial muscles.13–15 From a scientific point of view, the LCEs are fascinating because they uniquely hold high rubber–elasticity with their orientational order and reflect in the shape anisotropy of polymer strands in the network. As a result, they show electro-mechanical and electro-optical properties that can potentially surpass those of conventional systems. In addition, cholesteric LCEs also show piezoelectricity,16–22 and have the potential to act as a device that transforms a mechanical signal into an electric signal when the stress is applied parallel to the cholesteric helix.
Menthol has been used as a non-mesogenic monomer to synthesize side chain chiral LC copolymers.23–27 However, to the best of our knowledge, no research on LC compounds derived from menthol has been reported. Liu et al. reported a series of novel monomers and polymers containing menthyl groups.27 Although these chiral monomers and polymers contained two or three phenyl rings, their mesogenic cores were directly linked to terminal menthyl groups, and the existence of the bulky steric menthyl groups prevented the orientation of the LC molecule, so they showed no mesophase. However, recent research by the present authors showed that the compounds with three phenyl rings and terminal menthyl groups could form an LC phase when a flexible linkage chain was inserted between the mesogenic core and the menthyl fragments by reducing the steric effect.28
In the present paper, the synthesis and phase behavior of new chiral LC monomer and cholesteric elastomers derived from menthol will be investigated. The influence of the content of the crosslinking units on the phase behavior of the elastomers will be also discussed in detail.
Experimental section
Materials
All chemicals were obtained from the indicated sources and used as received. l-Menthol was obtained from Shanghai Kabo Chemical Co, chloroacetic acid from Tianjin Bodi Chemical Co., 4-hydroxybenzoic acid from Shanghai Wulian Chemical Plant, ethylene chlorohydrin from Tianjin Dagu Chemical Plant, undec-10-enoic acid from Beijing Jinlong chemical Reagent Co., 4, 4′-dihydroxybiphenyl and poly(methylhydro)siloxane (PMHS, DP = 35) from Aldrich.
Characterization
FT-IR spectra were recorded on a PerkinElmer spectrum One (B) spectrometer. 1H-NMR spectra were obtained with a Bruker ARX400 spectrometer. The samples were measured with tetramethylene sulfone (TMS) as internal standard in chloroform. The elemental analyses were carried out with an Elementar Vario EL III. The texture was observed with a Leica DMRX polarizing optical microscope (POM) equipped with a Linkam THMSE-600 cool and hot stage. The selective reflection wavelength was measured using a PerkinElmer 950 UV/Vis/NIR spectrometer with hot stage. The phase behavior was determined with a Netzsch DSC 204 equipped with a cooling system. The heating and cooling rates were 10°C min−1. The thermal stability of the elastomers under nitrogen atmosphere was measured with a Netzsch TGA 209C. The heating rates were 20°C min−1. X-ray diffraction (XRD) patterns of the elastomers were recorded with a nickel-filtered Cu-Kα (λ = 1.542 Å) radiation with a DMAX-3A Rigaku powder diffractometer. The instrument was operated at a voltage of 30 kV and a filament current of 30 mA. Radial scans of intensity vs. diffraction angle (2θ) were recorded in the range of 5~35°, and the scanning rate was 4° min−1.
Synthesis of the intermediate compounds
The synthetic route of the intermediate compounds is summarized in Scheme 1. Menthyloxyacetic acid (1), 4-menthyloxyacetoxybenzoic acid (3), 4-(2-hydroxyethoxy)benzoic acid (5), and4-(2-undec-10-enoyloxyethoxy)- benzoic acid (6) were synthesized according to the method reported by Hu.28–30
Synthetic route of the intermediate compounds 1−10.
4-(2-Undec-10-enoyloxyethoxy)benzoyl chloride was prepared by reacting 6 with excess thionyl chloride. The acid chloride obtained (11.0 g, 0.03 mol) was added dropwise to a solution of 4,4′-dihydroxybiphenyl (18.6 g, 0.1 mol) in 150 mL of tetrahydrofuran (THF) and 2.5 mL of pyridine under quickly stirring. The mixture was reacted for 6 h at room temperature, and for 8 h at 65°C. After the reaction mixture was concentrated, the crude product was precipitated by adding ice-water to the residue, and washed with 1.5% sodium hydroxide solution, then neutralized with diluted chlorhydric acid. The solid 7 was obtained by recrystallization from ethanol/acetone (3:1). Yield: 45%, mp: 154°C.
The synthetic route of the monomer 4-(menthyloxyacetoxybenzoyloxy)-biphenyl-4′-(2-(undec-10-enoyloxy)ethoxy)benzoate (ML) is outlined in Scheme 2. The compound 4 was prepared according to the reported literature.29 The acid chloride obtained (2.6 g, 0.02 mol), dissolved in 5 mL of chloroform, was added dropwise to a stirred solution containing the compound 7 (10.3 g, 0.02 mol) in 100 mL of chloroform and 2 mL of pyridine. The mixture was reacted at room temperature for 2 h and refluxed for 24 h, cooled to room temperature, filtered, and then concentrated. The crude product was precipitated by adding methanol to the filtrate, and purified by column chromatography (silica gel, dichloromethane).Yield: 55%. mp: 108°C. −22.6° (c = 0.2, chloroform).
Synthetic route of the monomer ML and crosslinking agent CA.
Elem. anal. calcd for C51H60O10: C, 73.53%; H, 7.26%. Found: C, 74.05%; H 7.38%.
Synthesis of the crosslinking agent
The synthetic route of the crosslinking agent 4-(undec-10-enoyloxy)biphenyl-4′(2-(undec-10-enoyloxy)ethoxy)benzoate (CA) is shown in Scheme 2. Undec-10-enoic acid (2.21 g, 12 mmol), N, N′-dicyclohexyl carbodiimide (DCC) (2.47 g, 12 mmol) and N, N′-dimethylaminopyridine (DMAP) (0.12 g, 1 mmol) were dissolved in 30 mL of dichloromethane. The compound 7 (5.16 g, 10 mmol), dissolved in 30 mL of dry dichloromethane at 30°C, were added to the above-mentioned solution. The reaction mixture was stirred for 30 h at 30°C. The resulting solution was washed with 10 mL of water, stirred for 20 min, and filtered. After removing the water, the organic layer was dried with anhydrous magnesium sulfate, and evaporated to dryness. The crude product was purified by column chromatography (silica gel, dichloromethane). White solid was obtained. Yield: 67%. mp: 79°C.
Elem. anal. calcd for C43H54O7: C, 75.63%; H, 7.97%. Found: C, 75.53%; H 8.09%.
Synthesis of the polymer networks
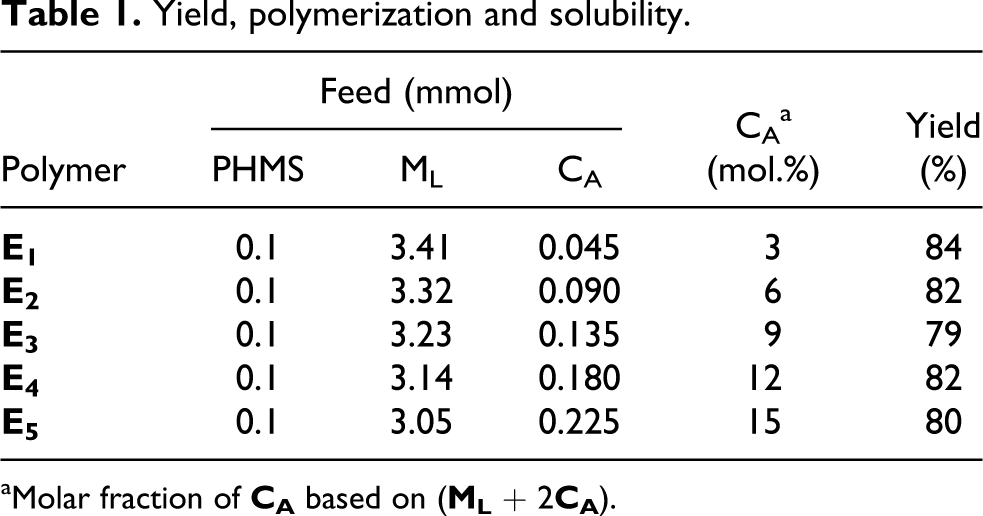
For the synthesis of the polymer networks E1−E5, the same method was adopted. The feed and polymerization of ML, CA, and PMHS were shown in Table 1. The reaction mixture, dissolved in dry toluene, was heated to 75°C under nitrogen and anhydrous conditions, and 2 mL of THF solution with the H2PtCl6 catalyst (5 mg mL−1) was injected into the mixture with a syringe. When the reaction was complete, the polymer networks were obtained by precipitation from toluene solution into methanol, purified by several filtrations from hot ethanol, and then dried in a vacuum.
Yield, polymerization and solubility.
Feed (mmol)
CAa
Yield
Polymer
PHMS
ML
CA
(mol.%)
(%)
E1
0.1
3.41
0.045
3
84
E2
0.1
3.32
0.090
6
82
E3
0.1
3.23
0.135
9
79
E4
0.1
3.14
0.180
12
82
E5
0.1
3.05
0.225
15
80
aMolar fraction of CA based on (ML + 2CA).
IR (KBr, cm−1): 2925, 2855 (−CH3, −CH2−); 1781, 1736 (C = O); 1606, 1494 (Ar−); 1280−1000 (Si−O−Si, C−Si and C−O−C).
Results and discussion
Structure characterization
The chemical structures of ML, CA and E1−E5 were characterized with FT-IR or 1H-NMR. IR spectra of ML and CAshowed characteristic stretching bands at 1781 cm−1 attributed to ester C = O in menthyloxyacetate, 1735 cm−1 attributed to ester C = O in undecylenate and substituted benzoate, 1641 cm-1 attributed to olefinic C = C, and 1606−1495 cm−1 attributed to aromatic C = C. 1H-NMR spectra of ML and CA showed multiplet at the chemical shift values nearby 0.83−4.65, 4.93−5.90, and 7.00−8.28 ppm corresponding to methyl and methylene protons, olefinic protons, and aromatic protons, respectively. The spectra suggested that the purity of ML and CA was high, which could be confirmed by the elemental analyses.
The elastomers E1−E5 were prepared by a one-step hydrosilylation reaction between Si-H groups of PMHS and olefinic C = C of ML and CA in dry toluene, using hexchloroplatinate hydrate as catalyst at 75°C. The progress of the hydrosilylation reaction, monitored by the Si−H stretch intensity, went to completion, as indicated by IR when the complete disappearance of Si–H stretching band at 2166 cm−1 and olefinic C = C stretching band at 1641 cm−1. In addition, characteristic Si−C bands at about 1262 and 783 cm−1, and Si−O−Si bands at about 1166, 1115 and 1025 cm−1 could been seen.
Polarizing optical microscopy analysis
To observe and record the phase behavior and optical textures of ML, CA and E1−E5, a POM with hot stage was used. The POM observations showed that ML revealed an enantiotropic chiral smectic C (SC*) phase and cholesteric phase. When ML was heated to 105°C, the sample started to melt, and a broken fan-shaped texture of a SC* phase gradually appeared. As heating continued to 150°C, the oily-streak texture of a cholesteric phase occurred, and subsequently disappeared at 180°C. When the isotropic state was cooled to 171°C, the focal conic texture occurred, at the same time, if a mechanical field was superimposed on the sample, for example, a slight shearing the melt could cause macroscopic orientation of the domains, and exhibited the property of selective reflection of light, which is typical of a cholesteric LC compound. As cooling continued to 140°C, the broken fan-shaped texture appeared again, and the sample crystallized at 88°C. In addition, on cooling the melt sample, the platelet texture of a cubic blue phase was first seen. As known, the blue phase reveals different color corresponding to different lattice planes, which shows Bragg scattering at different wavelengths, because the condition for constructive interference depends on the distance between lattice planes and their orientation with respect to the direction of incident light. The optical textures of MLare shown in Figure 1.
Optical textures of ML (200×): (a) oily streak texture of cholesteric phase on heating to 174°C; (b) platelet texture of a cubic BP on cooling to 172°C; (c) focal conic texture of cholesteric phase on cooling to 165°C; (d) broken fan-shaped texture of SC* phase on cooling to 116°C.
When the reflected wavelength lies in the visible range of the spectrum, both SC* and cholesteric phases can exhibit brilliant colors. Due to the angular dependence of the reflection conditions, the wavelength of the selective reflection of light λ obeys the Bragg condition:
where is the average index, P is the pitch of SC* or cholesteric phase, defined as the spatial distance over which the director rotates 360°, and θ is the incidence angle. When the normal incidence occurs, θ = 0o, a maximun wavelength λm is described as f0llows:
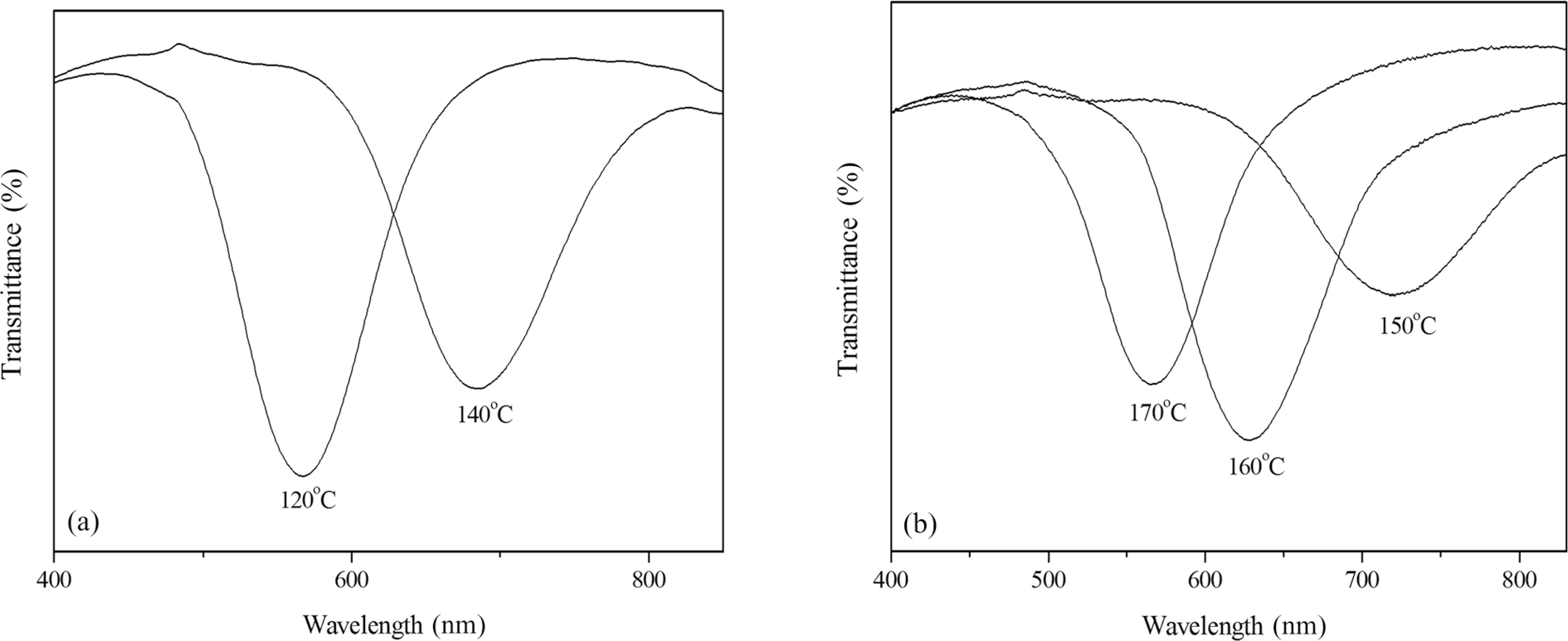
It is known that dP/dT > 0 for the SC* phase, and dP/dT < 0 for cholesteric phase, so the λm is temperature dependent. To describe the relationships of the λm and temperature, the λm of MLwas measured with UV/Vis/NIR spectra with hot stage. Figure 2 shows UV/Vis spectra of ML with temperature. It was clearly seen that the λm increased from 566 nm at 120°C to 684 nm at 140°C at SC* phase, while the λm decreased from 723 nm at 150°C to 562 nm at 170°C at cholesteric phase, which indicated the selective reflection shifted to the long wavelength region (red shift) at SC* phase, and to the short wavelength region (blue shift) at cholesteric phase with increasing temperature.
UV/Vis spectra of ML at SC*: (a) and cholesteric (b) phase temperature ranges.
The crosslinking agent CA exhibited an enantiotropic smectic A (SA) phase and nematic phase. When CA was heated to 81°C, the fan-shaped texture of a SA phase appeared at 85°C; the schlieren texture of a nematic phase was seen at 137°C. The optical textures of CAare shown in Figure 3.
Optical textures of CA (200×): (a) fan-shaped texture of SA phase on heating to 85°C; (b) schlieren texture of nematic phase on heating to 135°C.
Because of the copolymerization of the chiral monomer and nematic crosslinking agent, the elastomers E1−E4 showed cholesteric phase. The POM observation showed that E1−E3 exhibited color Grandjean texture and oily streak texture of a cholesteric phase. However, the texture of E4 was not typical, stress-induced birefringence could be seen, which is similar to the result described by Mitchell.31E5 showed no texture because of higher crosslinked density, this is consistent with the differential scanning calorimetry (DSC) result. The optical textures of E2, as an example, are shown in Figure 4.
Optical textures of E2 (200×): (a) color texture of cholesteric phase at 185°C; (b) oily streak texture of cholesteric phase at 219°C.
Differential scanning calorimetry analysis
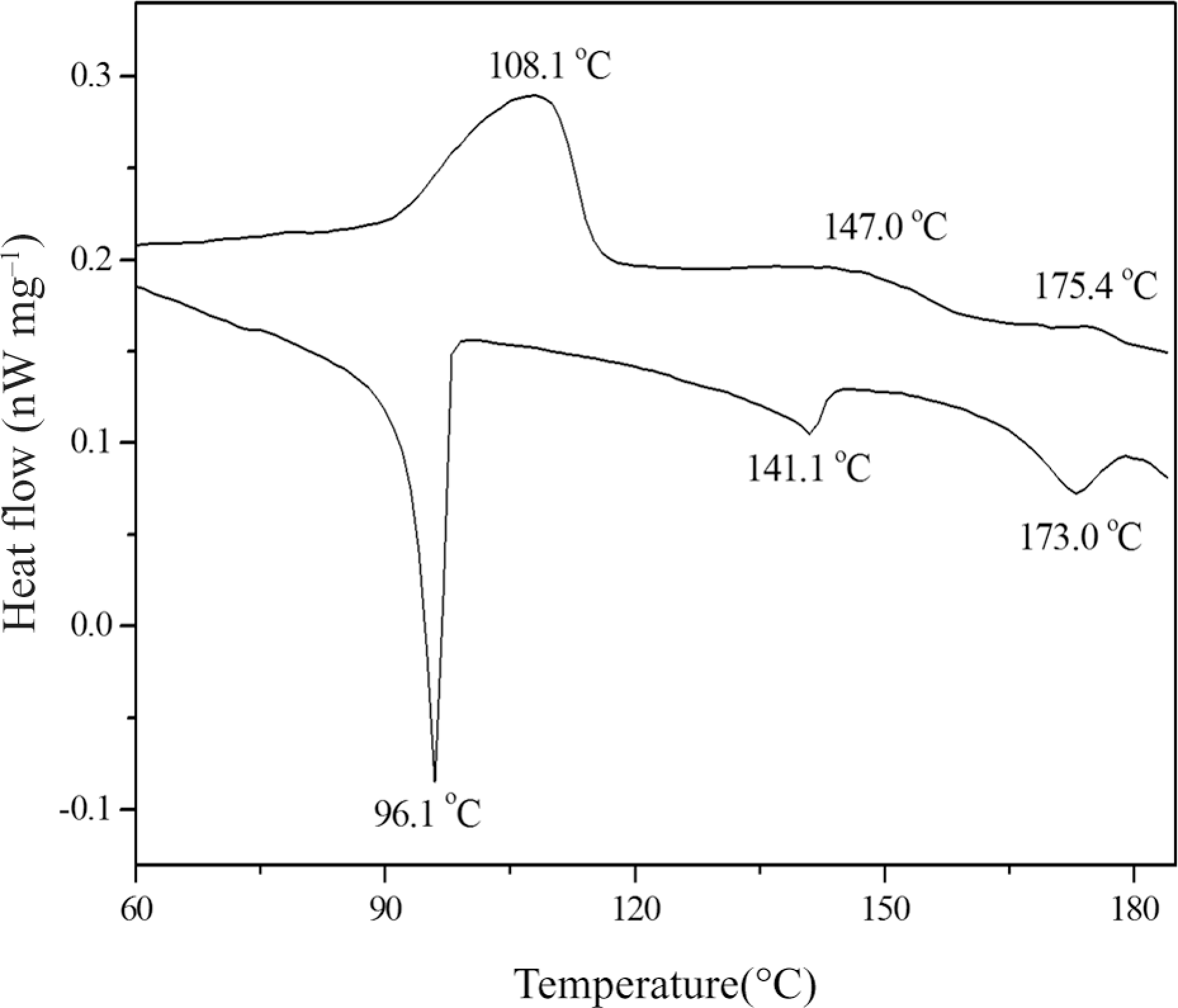
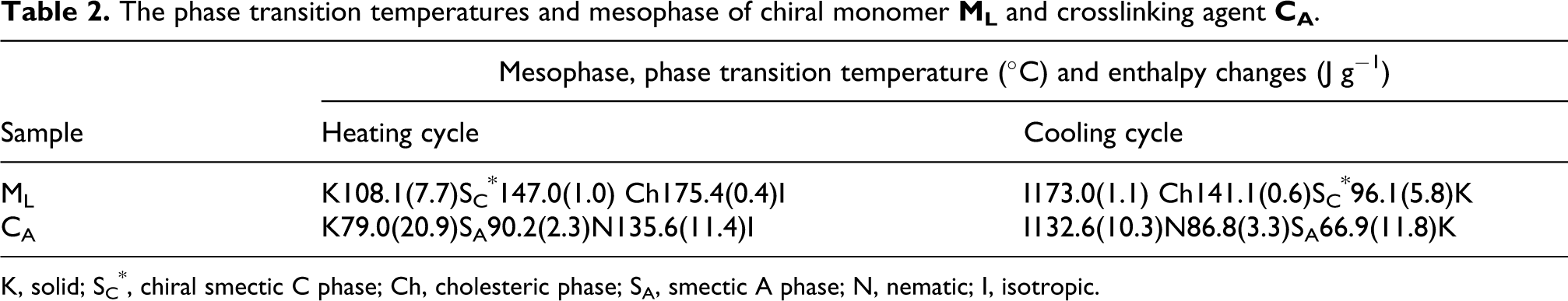
The thermal behavior of ML, CA and E1−E5 was studied with DSC. Their phase transition temperatures and corresponding enthalpy changes, obtained on the second heating and cooling scans, are summarized in Tables 2 and 3. Representative DSC curves of ML are shown in Figure 5.
DSC curves of ML.
The phase transition temperatures and mesophase of chiral monomer ML and crosslinking agent CA.
Sample
Mesophase, phase transition temperature (°C) and enthalpy changes (J g−1)
Heating cycle
Cooling cycle
ML
K108.1(7.7)SC*147.0(1.0) Ch175.4(0.4)I
I173.0(1.1) Ch141.1(0.6)SC*96.1(5.8)K
CA
K79.0(20.9)SA90.2(2.3)N135.6(11.4)I
I132.6(10.3)N86.8(3.3)SA66.9(11.8)K
K, solid; SC*, chiral smectic C phase; Ch, cholesteric phase; SA, smectic A phase; N, nematic; I, isotropic.
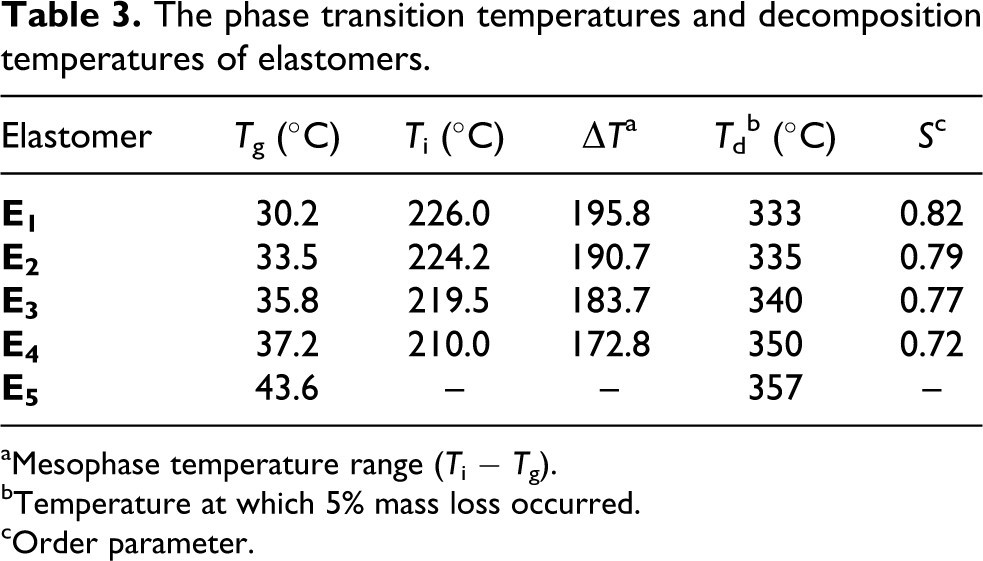
The phase transition temperatures and decomposition temperatures of elastomers.
Elastomer
Tg (°C)
Ti (°C)
▵Ta
Tdb (°C)
Sc
E1
30.2
226.0
195.8
333
0.82
E2
33.5
224.2
190.7
335
0.79
E3
35.8
219.5
183.7
340
0.77
E4
37.2
210.0
172.8
350
0.72
E5
43.6
–
–
357
–
aMesophase temperature range (Ti − Tg).
bTemperature at which 5% mass loss occurred.
cOrder parameter.
DSC curves of ML showed three endothermic peaks, which represented a melting transition at 108.1°C, a SC* to cholesteric phase transition at 147.0°C, and a cholesteric to isotropic phase transition at 175.4°C, respectively. On cooling, three exothermic peaks were seen, which represented an isotropic to cholesteric phase transition at 173.0°C, a cholesteric to SC* phase transition at 141.1°C, and a SC* to crystallization transition at 96.1°C, respectively.
DSC curves of CAshowed a melting transition at 79.0°C, a SA to nematic phase transition at 90.2°C, and a nematic to isotropic phase transition at 135.6°C, respectively. On cooling, three exothermic peaks represented an isotropic to nematic phase transition at 132.6°C, a nematic to SA phase transition at 86.8°C, and a SA to crystallization transition at 66.9°C, respectively.
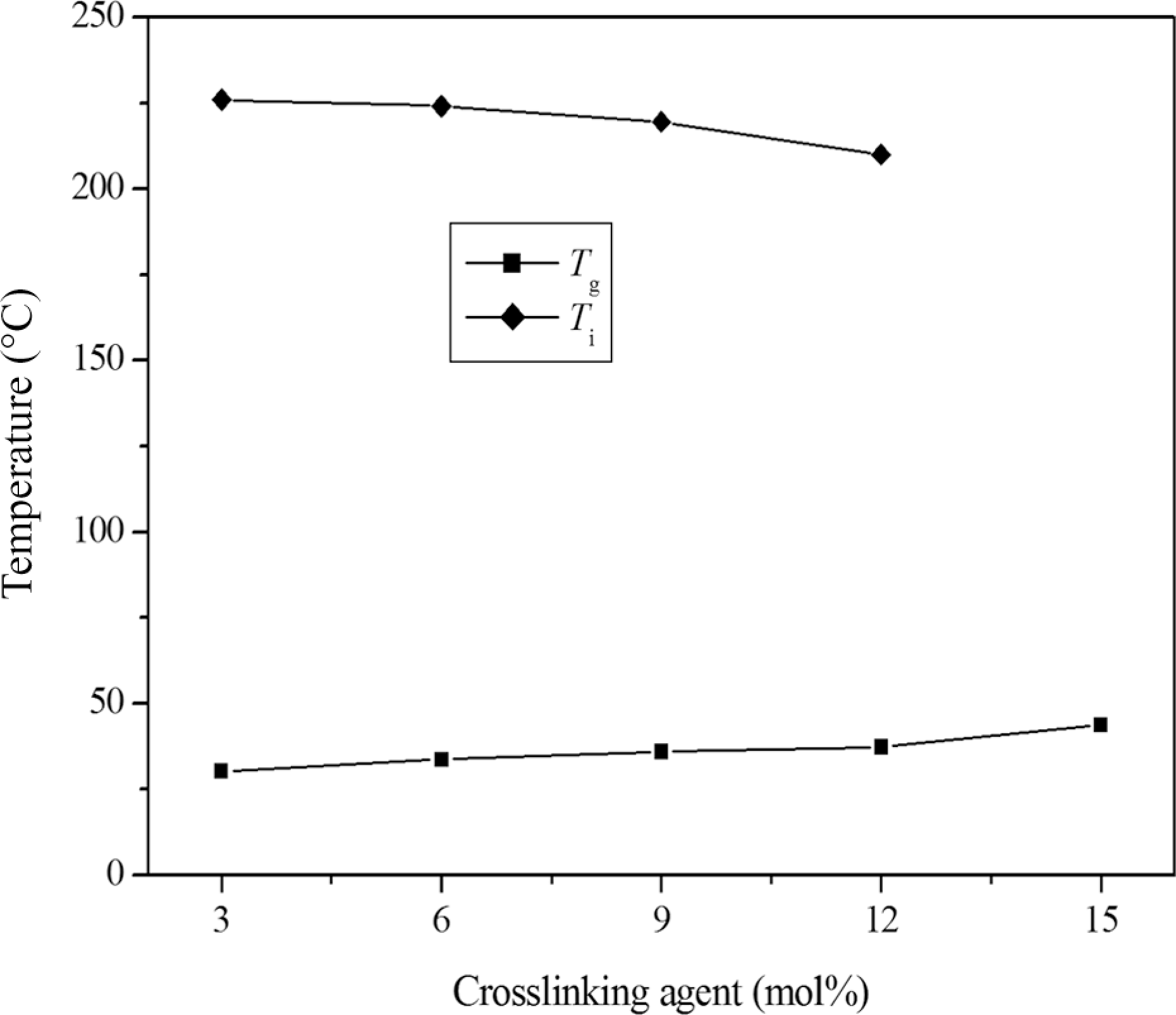
DSC heating curves of E1−E4 showed a glass transition at low temperature and a LC phase to isotropic transition at high temperature, which indicated that low crosslink density did not significantly affect the phase behavior of the elastomers, and reversible LC to isotropic phase transition could be observed because of enough molecular motion. In contrast, higher crosslink density had a strong influence on the phase behavior, it could make LC phase disappear due to the disturbance of the orientational-order. Therefore, E5 only showed a glass transition, this indicated that the LC properties disappeared when the content of crosslinking units was greater than 12 mol.%. Figure 6 shows the effect of crosslinking unit content on phase transition temperatures of the elastomers.
The effect of crosslinking unit content on phase transition temperatures of the elastomers.
It is known that the chemical crosslinking imposes additional constraints on the motion of chain segments, causes free volume to reduce, and leads to an increase in the glass transition temperature (Tg). Taking the crosslinking effect into account, the Tg is given by
where Tg and Tgo are the glass transition temperatures of the crosslinked and uncrosslinked polymers, Kx is a constant, and ρx is the crosslink density. It was clearly seen that the Tg increased from 30.2°C for E1 to 43.6°C for E5 when the content of crosslinking unit increased from 3 to 15 mol.%.
Similar to the Tg, the chemical crosslinking also affected the isotropic or clearing temperature (Ti) of the LCEs. On one hand, the flexible crosslinking chains acted as diluent and led to a decrease in Ti; on the other hand, the chemical crosslinking could prevent the motion and orientation of the mesogenic molecule in the vicinity of the crosslinking sites and did not favor the formation of the mesogenic orientational-order in the networks. So the Ti of the LCEs firstly decreased, and then disappeared with increasing crosslink density. When the content of the crosslinking unit increased from 3 to 12 mol.%, the corresponding Ti decreased from 226.0°C of E1 to 210.0°C of E4. In addition, E1−E4 showed wide mesophase temperature ranges (▵T), and ▵T decreased from 195.8°C of E1 to 172.8°C of E4 because the Ti decreased and the Tg increased.
Thermogravimetric analysis analysis
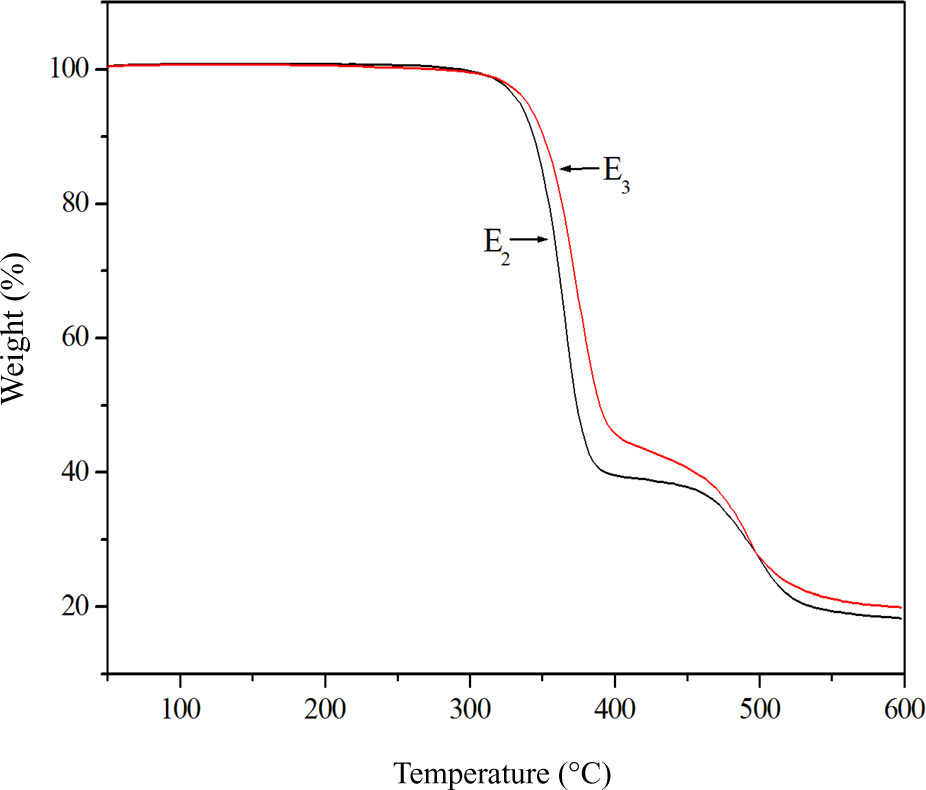
The thermal stabilities of E1−E5 were detected with TGA. Figure 7 shows representative TGA curves of E1 and E3. The corresponding data are summarized in Table 3. According to Figure 7, the main thermal decomposition appeared at the two temperature ranges. The weight was reduced by 45–60% when the temperature increased from 320 to 400°C; this was due to the thermal decomposition of the mesogenic unit and crosslinking unit. For the second temperature range from 470 to 520°C, the weight decreased by 20%. Moreover, the temperatures at which 5% weight loss occurred (Td) of E1−E5increased with increasing the concentration of the crosslinking units. In addition, the Td of E1−E5was greater than 330°C, this indicated that all the elastomers had excellent thermal stabilities.
TGA curves of E1 and E3.
X-ray diffraction analysis
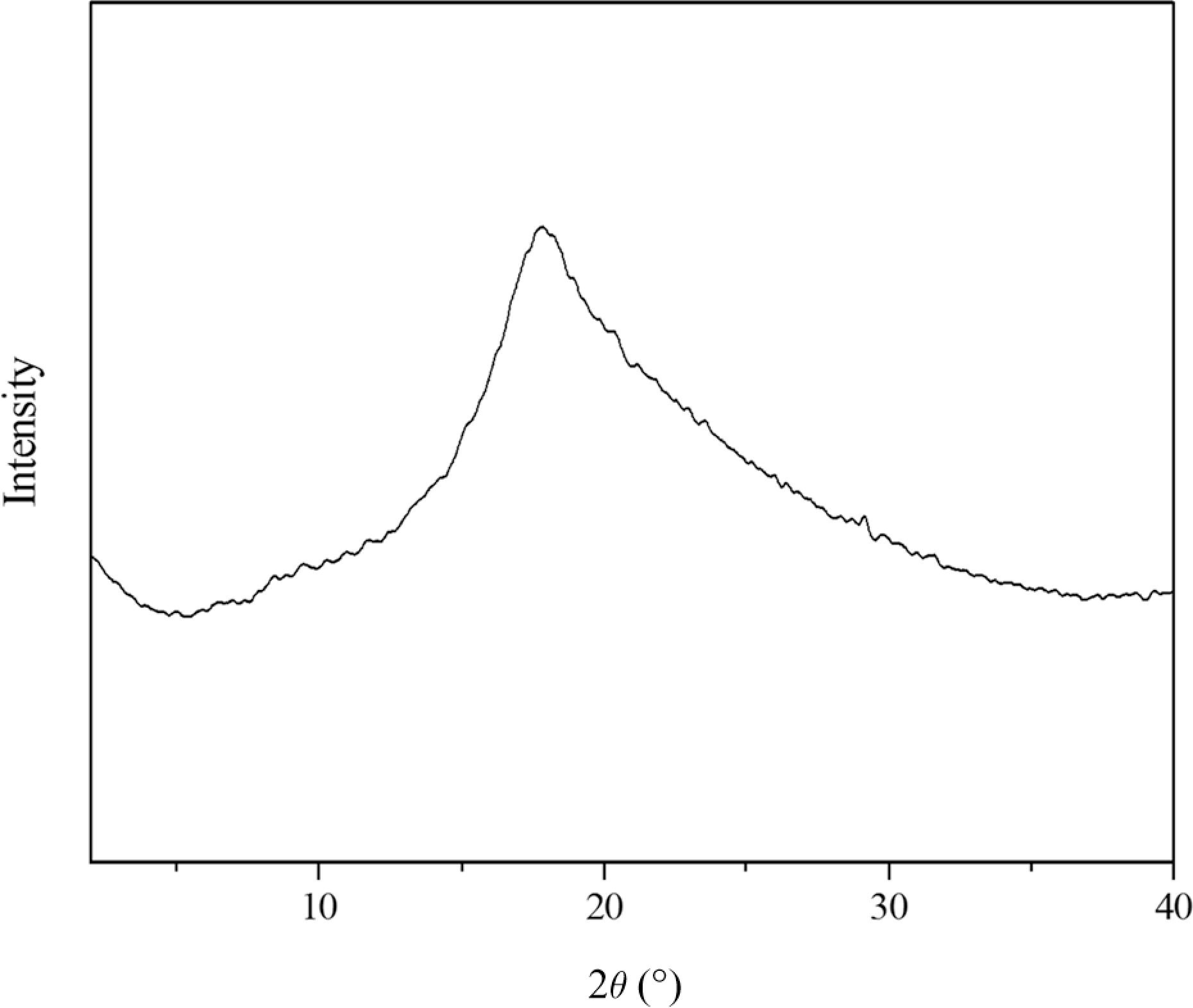
To obtain more detailed information on the LC structure, XRD was used. In general, a sharp and strong peak associated with the smectic layers at small angle (1° < 2θ < 4°) and a broad peak associated with lateral packing (the molecular arrangement of the mesogenic side groups) at wide angle (2θ = 18–20°) can be observed for smectic structure. No peak appears at a small angle, and only a broad peak occurs at a wide angle for nematic and cholesteric structure. For E1−E4, XRD could confirm the presence of the cholesteric phase, which is in agreement with POM observation results. A sharp peak associated with the smectic layers at small angle did not appear, whereas a broad peak were observed at 2θ = 18o. Moreover, the broad peaks were more and more diffuse, and peak intensity reduced with increasing crosslink density, this indicated the decrease of the LC order. Figure 8 shows XRD curve of E3 at 90°C as an example.
XRD curve of E3.
Nearly perfect orientations of the phase structure for the cholesteric elastomers can be obtained by applying external mechanical fields or the so-called anisotropic deswelling method.32,33 For the oriented elastomers, the average degree of orientation of the LC domains can be represented by the orientational order parameter S, which is defined according to the following equation:
where φ is the angle between a particle axis and the director that is the vector that specifies the average alignment direction of the particles (S = 0 for an isotropic system and S = 1 for a perfect alignment). The order parameters of the LCEs are determined by X-ray measurements, which are shown in Table 3.
Conclusions
The synthesis and phase behavior of a new chiral mesogenic monomer ML derived from menthol, crosslinking agent CA and the corresponding elastomers E1−E5 were investigated. ML exhibited the broken fan-shaped of a SC* phase, platelet texture of a cubic blue phase and oily streak texture or focal conic texture of a cholesteric phase. The λm increased at SC* phase range, and decreased at cholesteric phase range with increasing temperature. CA exhibited typical enantiotropic fan-shaped texture of a SA phase and the schlieren texture of a nematic phase. E1−E4 showed the cholesteric phase. When the content of crosslinking unit increased from 2 to 12 mol.%, the Tg increased from 30.2°C for E1 to 37.2°C for E4, while the Ti decreased from 226.0 to 210.0°C. In addition, all the elastomers displayed very good thermal stability.
Footnotes
Acknowledgement
This study was supported by Science and Technology Bureau of Shenyang, and Fundamental Research Funds for the Central Universities (N110405006, N110705001 and N110405001).
References
1.
OrtizCOberCKKramerEJ. Stress relaxation of a main-chain, smectic, polydomain liquid crystalline elastomer. Polymer1998; 39: 3713–3718.
2.
HsuCSChenHL. Preparation of liquid-crystal thermosets: in situ photopolymerization of oriented liquid-crystal diacrylates. J Polym Sci Part A: Polym Chem1999; 37: 3929–3935.
3.
LiMHuZJChenG. Phase behavior of side-chain liquid-crystalline elastomers and their precursors containing para-nitroazobenzene. J Appl Polym Sci2003; 88: 2275–2279.
4.
AraiYOUrayamaKKohjiyaS. Role of network nematicity in swelling and phase equilibria of polymer networks in nematic solvents. Polymer2004; 45: 5127–5135.
5.
SaikrasunSBualek-LimcharoenSKohjiyaS. Anisotropic mechanical properties of thermoplastic elastomers in situ reinforced with thermotropic liquid-crystalline polymer fibers revealed by biaxial deformations. J Polym Sci Part A: Polym Phys2005; 43: 135–144.
6.
HuJSZhangBYZhouAJ. Side-chain cholesteric liquid crystalline elastomers derived from a mesogenic crosslinking agent: synthesis and mesomorphic properties. Eur Polym J2006; 42: 2849–2858.
7.
BeyerPTerentjevEMZentelR. Monodomain liquid crystal main chain elastomers by photocrosslinking. Macromol Rapid Commun2007; 28: 1485–1490.
8.
BalamuruganRKannanP. Synthesis and properties of a liquid crystalline thermoset epoxy resin containing 1,3,4-oxadiazole groups. High Perform Polym2009; 21: 251–264.
9.
IqbalMDingemansTJ. Synthesis, characterization and properties of branched all-aromatic liquid crystal thermosets. High Perform Polym2010; 22: 891–904.
10.
VerduzcoRLuchettePHongSH. Bent-ore liquid crystal elastomers. J Mater Chem2010; 20: 8488–8495.
11.
OhmCHaberkornNTheatoP. Template-based fabrication of nanometer-scaled actuators from liquid-crystalline elastomers. Small2011; 7: 194–198.
12.
Sanchez-FerrerAFinkelmannH. Polydomain-monodomain orientational process in smectic-c main-chain liquid-crystalline elastomers. Macromol Rapid Comm2011; 32: 309–315.
13.
PapadopoulosPHeinzePFinkelmannH. Electromechanical properties of smectic c* liquid crystal elastomers under shear. Macromolecules2010; 43: 6666–6670.
14.
YangHBuguinATaulemesseJMK. Micron-sized main-chain liquid crystalline elastomer actuators with ultralarge amplitude contractions. J Am Chem Soc2009; 131: 15000–15004.
15.
OhmCBrehmerMZentelR. Liquid crystalline elastomers as actuators and sensors. Adv Mater2010; 22: 3366–3387.
AltomareAAndruzziLCiardelliF. Methacrylic polymers containing permanent dipole azobenzene chromophores spaced from the main chain: synthesis and characterization. Polym Int1998; 47: 419–427.
25.
BobrovskyAYBoikoNIShibaevVP. The induced SA phase in new menthyl-containing copolymers. Macromolecules1998; 31: 5800–5804.
26.
BobrovskyAYShibaevVP. Chiral nematic polymer mixture containing crosslinker and photosensitive chiral dopant: new type of materials with tunable photo-optical properties. Adv Funct Mater2002; 12: 367–372.
27.
LiuJHYangPC. Synthesis and characterization of novel monomers and polymers containing chiral (-)-menthyl groups. Polymer2006; 47: 4925–4935.
28.
HuJSWeiKQZhangBY. Synthesis, structure and properties of new chiral liquid crystalline monomers and homopolysiloxanes containing menthyl groups. Liq Cryst2008; 35: 925–935.
29.
HuJSZhangBYJiaYG. Structures and properties of side-chain cholesteric liquid crystalline polyacrylates. Polym J2003; 35: 160–166.
30.
HuJSZhangBYZhouAJ. Preparation and phase behavior of side-chain cholesteric liquid-crystalline elastomers. J Polym Sci Part A: Polym Chem2005; 43: 3315–3323.
31.
MitchellGRDavisFJAshmanA. Structural studies of side-chain liquid crystal polymers and elastomers. Polymer1987; 28: 639–647.
32.
HiraokaKFinkelmannH. Uniform alignment of chiral smectic C elastomers induced by mechanical shear field. Macromol Rapid Commun2001; 22: 456–460.
33.
KimSTFinkelmannH. Cholesteric liquid single-crystal elastomers (LSCE) obtained by the anisotropic deswelling method., Macromol Rapid Commun2001; 22: 429–433.