
Abstract
In this study, azomethine polyphosphonates were synthesized by solution polycondensation of phenylphosphonic dichloride with various azomethine diols such as [4-(4-hydroxy phenyl) iminomethyl] phenol, [(4-(4-hydroxy-3-methoxy phenyl) iminomethyl)] phenol and [4-(4-hydroxy-3-ethoxy phenyl) iminomethyl] phenol using triethylamine catalyst at ambient temperature. The structure of the synthesized polymers was confirmed by Fourier transform infrared and 1H-, 13C- and 31P- nuclear magnetic resonance spectroscopic techniques. Thermal properties of the polymers were studied by thermogravimetric analysis (TGA) and differential scanning calorimetry under nitrogen atmosphere. The TGA data showed that the synthesized polyphosphonates produce high char yield at 600°C due to the presence of phosphorous atom in the polymer chain and hence have good flame-retardant properties. One of the synthesized polyphosphonate was blended with commercial diglycidyl ether of bisphenol-A (DGEBA) resin in various weight percentage and cured with commercial curing agent triethylene tetramine (TETA). The polyphosphonates-blended epoxy thermosets have tensile strength in the range of 5–41 MPa and the percentage of elongation at breaks was 4–18. It was found that the incorporation of polyphosphonates into epoxy thermoset decreased the tensile strength from 41 MPa to 5 MPa, whereas the elongation at break value increased with increase in the weight percentage of polyphosphonate. The influence of polyphosphonates on the flame retardancy of blended thermosets was examined by limiting oxygen index (LOI) and vertical burning (UL-94) tests and found that the polymer samples achieved an increased UL-94 rating and the LOI values were in the range of 24–26. Broido and Horowitz–Metzger methods have been used to study the thermal degradation kinetic parameters.
Keywords
Introduction
Polyphosphonates are the most promising flame-retardant materials which contribute towards the formation of thermally stable char layer and inhibit heat transfer. 1,2 Their products of decomposition are less toxic and corrosive and cause little damage to the environment during fire, hence phosphorous-containing
flame retardants received immense attention as additives in the polymer field. During polymerization, phosphorous becomes active in the condensed phase depending on the chemical nature and thermal stability of the host polymer matrices. 3,4 Other interesting features of phosphorous polymers are good adhesion to substrates, metal ion binding characteristics, increased polarity, plasticity and lubricant properties. 5 The development of flame-retardant polymers has been an important area of research over the last few years. The drive for new materials can be attributed to the need in aerospace industry and government regulations for fire safety. 6 –8 Polyphosphonates are important class of organophosphorus-based flame-retardant polymer additives. They not only display good flame retardancy but also possess attractive plasticizing properties. The reason for the use of polymeric or oligomeric additives, rather than conventional non-polymeric species, is that they show better resistance to extraction, migration and volatile-loss, thus making them more attractive.
Poly(azomethine)s system with conjugated –CH=N– bonds exhibit valuable properties mainly associated with the conjugated backbone and the presence of imine sites. 9 –11 The unique role of azomethine in conjugated polymers is connected with the presence of nitrogen atom in the azomethine group and its free electron pair, which may take part in the formation of complexes with electrophilic substances. 12 This provides the possibility for a wide range of applications of the polymers and for modifications of their properties. Polyazomethines are attractive high-performance polymers 13,14 due to their high thermal stability, 15,16 excellent mechanical strength 17 and liquid crystalline property 18 . These polymers are generally difficult to process due to their high melting point and poor solubility. 12,19 Aromatic azomethines are stable in the reaction medium in which they are produced, as well as after isolation. The processibility of polyazomethines without sacrificing high thermal stability can be improved by the introduction of various substituted aromatic ring into the polymer backbone. 20 Phenyl phosphonic dichloride is a typical intermediate to prepare main chain phosphorus-containing material due to its two highly reactive phosphoryl chlorides, which can easily react with the compounds containing hydroxyl or amino groups. 21,22 Introduction of phosphorous linkages into these azomethine polymers can also improve the solubility. Flame retardants are incorporated into polymer to achieve the desired fire resistance. Even though the presence of nitrogen compounds exhibits minor contribution to flame-retardant effects, public focus is much more due to its eco-friendly behaviour. Polymers containing both nitrogen and phosphorous exhibit unusual thermal properties such as flame retardancy and self-extinguishability. 23,24 These characteristics can be achieved by the incorporation of azomethine linkage into polyphosphates.
The present study deals with the synthesis of polyphosphonates by reacting phenyl phosphonic dichloride with various azomethine diols such as [4-(4-hydroxy phenyl) iminomethyl] phenol, [(4-(4-hydroxy-3-methoxy phenyl) iminomethyl)] phenol and [4-(4-hydroxy-3-ethoxy phenyl) iminomethyl] phenol by solution poly condensation technique. All the synthesized polyphosphonates were systematically characterized by spectroscopic techniques, and their thermal, flame-retardant and mechanical properties were investigated. A detailed thermogravimetric study was carried out using dynamic/non-isothermal methods in order to determine the activation energy (E a) and to select the most appropriate mechanism to describe the degradation behaviour. The value of E a for the decomposition of polyphosphonates in various steps and the regression coefficients have been calculated by employing the Broido and Horowitz–Metzger methods and the results have been compared.
Experimental
Materials
4-Hydroxy benzaldehyde and 4-hydroxy-3-methoxy benzaldehyde were obtained from S D Fine Chemicals (Mumbai, Maharashtra, India). 4-Amino phenol and tetrahydrofuran were obtained from Merck Chemicals (Mumbai, Maharashtra, India). All the other solvents used in the study were purified by the procedure reported in the literature. 25,26 3-Ethoxy-4-hydroxy benzaldehyde, phenyl phosphonic dichloride and triethylamine were procured from Sigma Aldrich (Munich, Germany) and used without further purification. Epoxy resin (LY556) and hardener (HY951) were procured from Covai Seeno Company (Coimbatore, Tamil Nadu, India).
Characterization
The solubility of the monomer and polymers was tested with 0.2–0.5 mg of substance in 5 mL of solvents at room temperature. Viscosity measurements were carried out with 0.3 g dL−1 of polymer solution in dimethylsulphoxide (DMSO) at room temperature using an Ostwald viscometer (Amit Enterprises, India). The infrared spectra were recorded on a Fourier transform spectrophotometer (Shimadzu, Japan) using potassium bromide (KBr) pellets. Ultraviolet (UV)-visible spectra were recorded in DMSO solution with Cary 60 UV-visible spectrophotometer (Agilent Technologies, Germany). 1H- and 13C-nuclear magnetic resonance (NMR) spectra were recorded on a 400-MHz NMR spectrometer (AV-III 400, Bruker, Billerica, Massachusetts, USA) using deuterated DMSO. The chemical shifts were measured using tetra methyl silane (TMS) as an internal standard. The elemental analysis was carried out using Vario EL III carbon, hydrogen, nitrogen and sulphur analyser (Elementar Analysensysteme, Hanau, Germany). The differential scanning calorimetry (DSC) analysis was carried out using Pyris 6 DSC (PerkinElmer, Waltham Massachusetts, USA) with an empty aluminium pan as a reference at a heating rate of 10°C min−1 under nitrogen atmosphere. The temperature and heat flow scale of the instrument were calibrated with pre-crimped indium and zinc as standard references. Thermogravimetric analysis (TGA) was performed on a thermal analyser (Seiko Instruments, SII Nanotechnology, Japan) under nitrogen atmosphere at a heating rate of 10°C min−1. The tensile strength of the specimen was measured using universal testing machine (FMI, TFT-50KN-D, Metest equipments, India) following ASTM D 638 standard specification with the sample size of 90 × 18 × 3 mm3. The flame retardancy of all the samples was characterized by the limiting oxygen index (LOI) and UL-94 vertical burning classification tests. The LOI of the specimen was determined using an LOI analyser (Dynisco, Franklin, Massachusetts, USA) according to ASTM D2863 standard specification with the sample size of 150 ± 1 × 6.5 ± 1 × 3 ± 0.1 mm3. The UL94 vertical burning classification test was accomplished using a burning chamber according to ANSI/UL-94-1985 with the specimen of 130 ± 1 × 13 ± 1 × 3.2 ± 0.1 mm3. The UL94 test results were classified by the burning ratings as V-0, V-1 and V-2. The tensile strength and flame retardancy were calculated by taking an average of five individual determinations.
Thermal degradation study
Thermal analysis methods are often used for the determination of degradation processes that take place during progressive heating of a polymeric material as well as for the evaluation of the kinetic parameters of each degradation step. There are several methods proposed by Friedman, F Chang, Kissinger, Flynn-wall, Broido, Horowitz–Metzger, Coats–Redfern, Van Krevelen, Ozawa and Vyazovkin for calculating kinetic parameters that depend not only on the experimental conditions but also on the mathematical treatment of the data. In this article, Broido and Horowitz–Metzger methods were employed to evaluate activation energy (E a) using single heating rate measurement without making any assumptions. The kinetic parameters were calculated according to linear regression method using the following kinetic equation
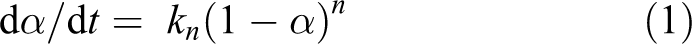
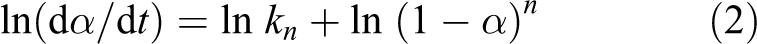
where α is the fraction of the sample weight at time ‘t’, kn is the specific rate with kinetic reaction order of ‘n’ and (1 − α) is the fraction of the numbers of initial molecules that are not yet decomposed.
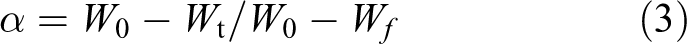
where W
0 is the initial sample weight, W
t is the sample weight at time t and W
f is the final sample weight. The reaction rates
Broido model
The activation energy (E a) associated with each stage of decomposition was also evaluated by the well-known Broido method. 28 The equation used for the calculation of E a is
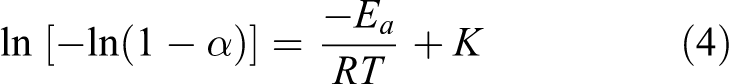
E
a was calculated from the plot of ln [−ln(1 − α)] versus
Horowitz and Metzger model
Horowitz and Metzger 29 have demonstrated a method to calculate the activation energy (E a) of polymeric substances. The equation used for calculation of E a is given below
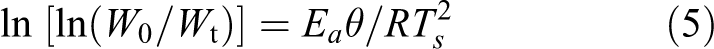
where θ = T − T s; W 0 is the initial weight, W t is the weight at any time ‘t’, T s is the differential thermogravimetric peak temperature and T is the temperature corresponding to particular weight loss. A plot of ln [ln (W 0/W t)] versus θ gives an excellent approximation to a straight line, and from the slope, E a was calculated.
Synthesis of monomers
Synthesis of 4-[(4-hydroxy phenyl)imino methyl] phenol
To a stirred solution of 4-aminophenol (0.1 mol) in hot ethanol, ethanolic solution of 4-hydroxy benzaldehyde (0.1 mol) was added dropwise. The mixture was stirred for 4 h at 70°C, cooled and the resultant solid precipitate was filtered, dried and recrystallized from ethanol (melting point: 214°C; yield: 80%). 30 The scheme of preparation is shown in Figure 1. Analysis calculated for C13H10O2N is as follows: C, 72.3; H, 4.69; N, 6.57%. Found: C, 68.01; H, 5.44; N, 6.07%. Fourier-transform infrared spectroscopy (FTIR; KBr, cm−1): 3372 (γ O–H), 1526(γ C=C, Ar), 1624(γ −CH=N) and 1236(γ C–O, phenol); 1H-NMR (DMSO-d 6, TMS, ppm): 6.77–7.74 (m, aromatic protons), 10.02 (s, 2H, OH protons) and 8.44 (s, −CH=N); and 13C-NMR (DMSO-d 6, TMS, ppm): 115.5–155.6 (aromatic carbons) and 160.05(–CH=N).
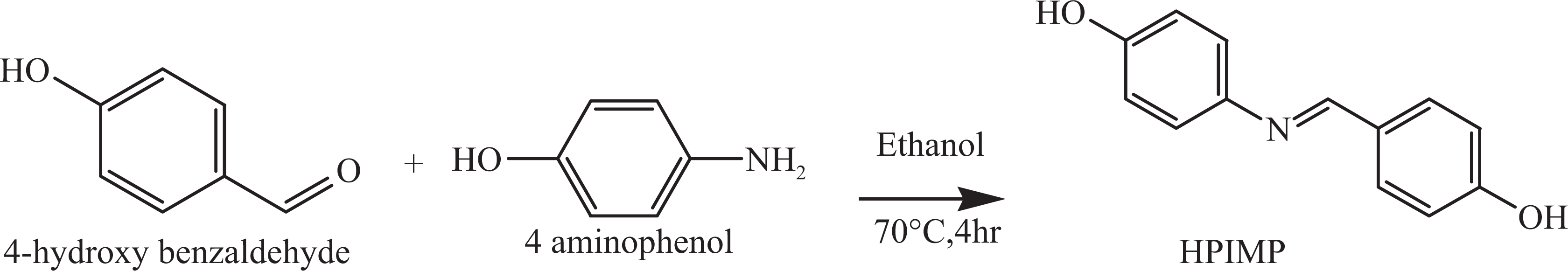
Synthesis of HPIMP. HPIMP: 4-[(4-hydroxy phenyl)imino methyl] phenol.
The monomers (4-(4-hydroxy-3-methoxy phenyl) iminomethyl) phenol (HMPIMP) and (4-(4-hydroxy-3-ethoxy phenyl) iminomethyl) phenol (HEPIMP) were synthesized from 4-hydroxy-3-methoxy benzaldehyde and 4-hydroxy-3-ethoxy benzaldehyde, respectively, by adopting the procedure followed for the synthesis of monomer, 4-[(4-hydroxy phenyl)imino methyl] phenol (HPIMP). IR and NMR spectral data of all the monomers are summarized in Table 2.
Synthesis of polyphosphonates
All the polyphosphonates were synthesized by the solution polycondensation technique using triethylamine catalyst 31 as shown in Figure 1. The procedure for the synthesis of P1 is described subsequently. The monomer HPIMP (0.005 mol) was dissolved in dry tetrahydrofuran (20 mL) and stirred for 5 min with the addition of triethylamine (0.010 mol). To this mixture, phenyl phosphonic dichloride (0.005 mol) in 20-mL tetrahydrofuran was added dropwise using an addition flask and the mixture was stirred vigorously for 4 h at ambient temperature. During this period, the viscosity of the solution increased rapidly and the polymer began to precipitate. The brownish yellow polymer precipitated was filtered, washed with acetone and then dried under vacuum for 24 h (yield: 79%). The scheme of preparation is shown in Figure 2.
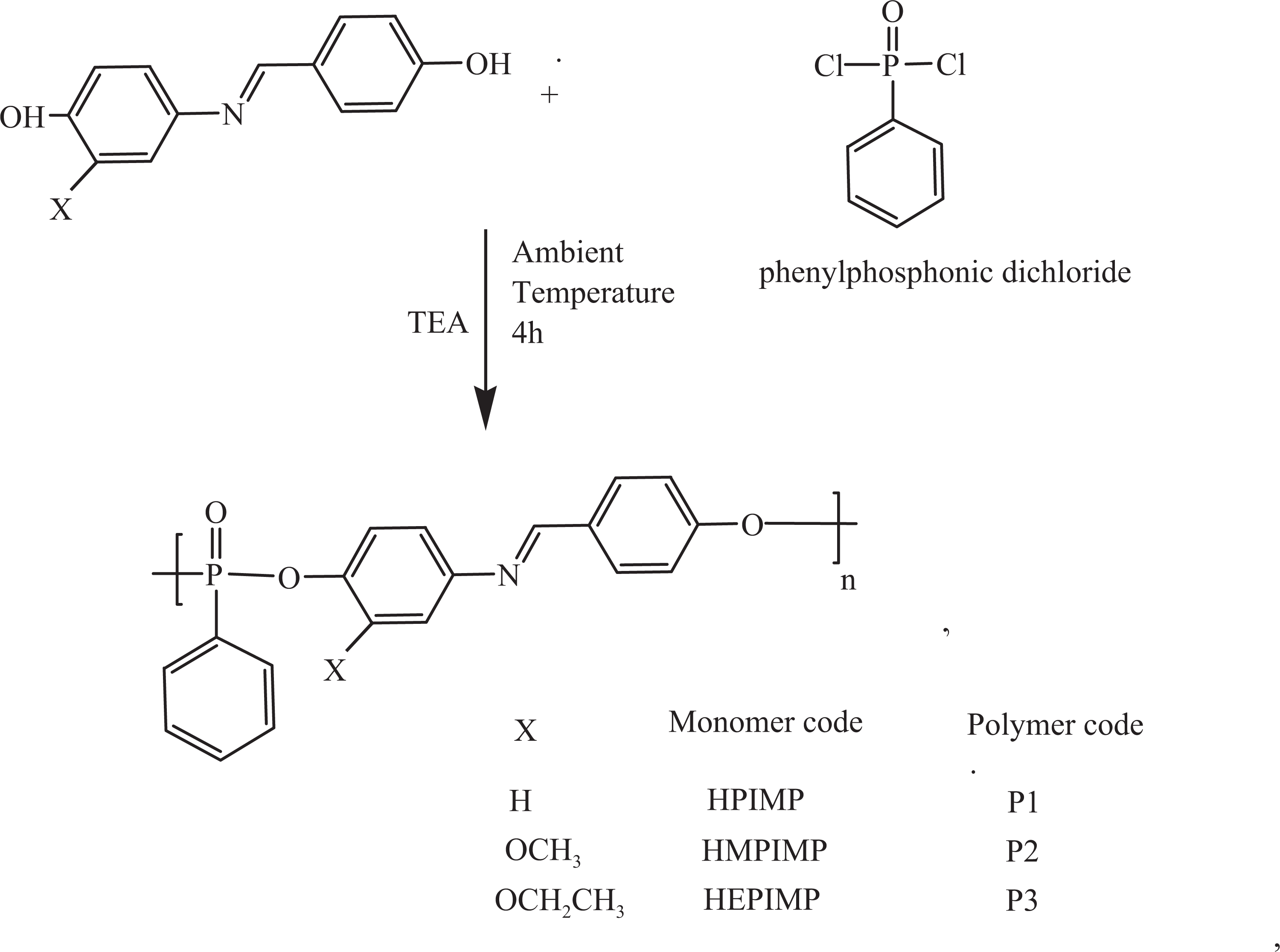
Synthesis of polyphosphonates.
FTIR (KBr, cm−1): 2982 (γ C–H), 1560 (γ C=C Ar), 1618 (γ –CH=N–), 1262 (γ –P=O) and 1156 (γ P–O–C Ar); 1H-NMR (DMSO-d 6, TMS, ppm): 6.79–7.80 (m, Ar) and 8.52 (s,–CH=N); 13C-NMR (DMSO-d 6, ppm): 191.2 (ester carbonyl carbon), 163.4 (CH=N carbon) and 115.7–156.8 (aromatic carbons); and 31P-NMR (DMSO, TMS, ppm): 12.22 ppm (aromatic phosphorous).
Polyphosphonates P2 and P3 were synthesized from HMPIMP and HEPIMP using similar procedure followed for the synthesis of polyphosphonate P1.
Tensile strength test
Tensile strength testing was carried out as per ASTM D 3039 on Fine Testing Machine (Model 6025UK) with 10 mm min−1 cross-head speed of 30 KN, using specimens fabricated by blending varying weight percentage of synthesized polyphosphonate P1 (1%, 3%, 5%, 7% and 9%) with commercial epoxy resin and cured with hardener. This was dried in a hot air oven at 70°C for 3 h. Five specimens of each sample were tested and the average was calculated.
Flammability test
The specimens for LOI and UL-94 vertical burning tests were prepared in accordance with the ASTM D2863. Synthesized polyphosphonate P1 was blended with commercial epoxy resin in varying weight percentage (1%, 3%, 5%, 7% and 9%) and cured with hardener. This was dried in a hot air oven at 70°C for 3 h.
Results and discussion
Synthesis and characterization
The aromatic azomethine containing polyphosphonates P1, P2 and P3 were prepared in good yield using solution polycondensation technique by reacting the synthesized bisphenol monomers with phenyl phosphonic dichloride. The yield, solubility and inherent viscosity data of the polyphosphonates are given in Table 1. It was found that all the polyphosphonates were soluble in dimethyl sulphoxide at room temperature and insoluble in aprotic solvents, such as dichloromethane, tetrahydrofuran and chloroform. The inherent viscosity of synthesized polymers was found to be in the range of 0.34–0.42 g dL−1. The structure of the synthesized polyphosphonates was confirmed by FTIR and NMR spectroscopic techniques. The FTIR spectrum of polymer P1 is shown in Figure 3. The absorption at around 3400 cm−1 is due to the presence of terminal phenolic –OH group of polymers. The characteristic absorption band at around 1262 cm−1 corresponds to the P=O stretching. 32 –34 The peaks at around 1156–1178 cm−1 and 928 cm−1 correspond to P–O–C aromatic stretching. The stretching frequency of –CH=N– of the polymer appeared in the region of 1618 cm−1. The 1H-NMR spectrum of polyester P1 is shown in Figure 5. In 1H-NMR, the signal at 10.4 ppm attributed to terminal –OH group of polymers. The aromatic protons appeared as a broad multiplet in the region of 6.79–7.80 ppm. The resonance peaks of imine, –CH=N–, protons appeared at 8.52 ppm and the methylene protons of diol monomers appeared in the region of 1.17–2.50 ppm. 35 The complex and wider aromatic proton signals of polymer P1 compared to the monomer HPIMP may be attributed to the polymer formation. The 13C-NMR spectrum of polyester P1 is shown in Figure 6. The ester carbonyl carbon peak appeared at 191.2 ppm. The aromatic carbons of phenyl ring resonated between 115.7 and 156.89 ppm. These chemical shift values of diol monomers are different from that of polymers. It confirms that both the phenolic –OH groups have taken part in the polymerization reaction. The peak observed at around 163.4 ppm is ascribed to –CH=N– carbon. The 31P-NMR spectrum of polymer P1 is shown in Figure 7. The signal centered at around 12.22 ppm corresponds to the aromatic phosphonate. 36,37 All these spectral results confirmed the formation of polyphosphonates.
Yield, solubility and inherent viscosity data of the polyphosphonates.

++: soluble at room temperature; −−: insoluble even on heating.
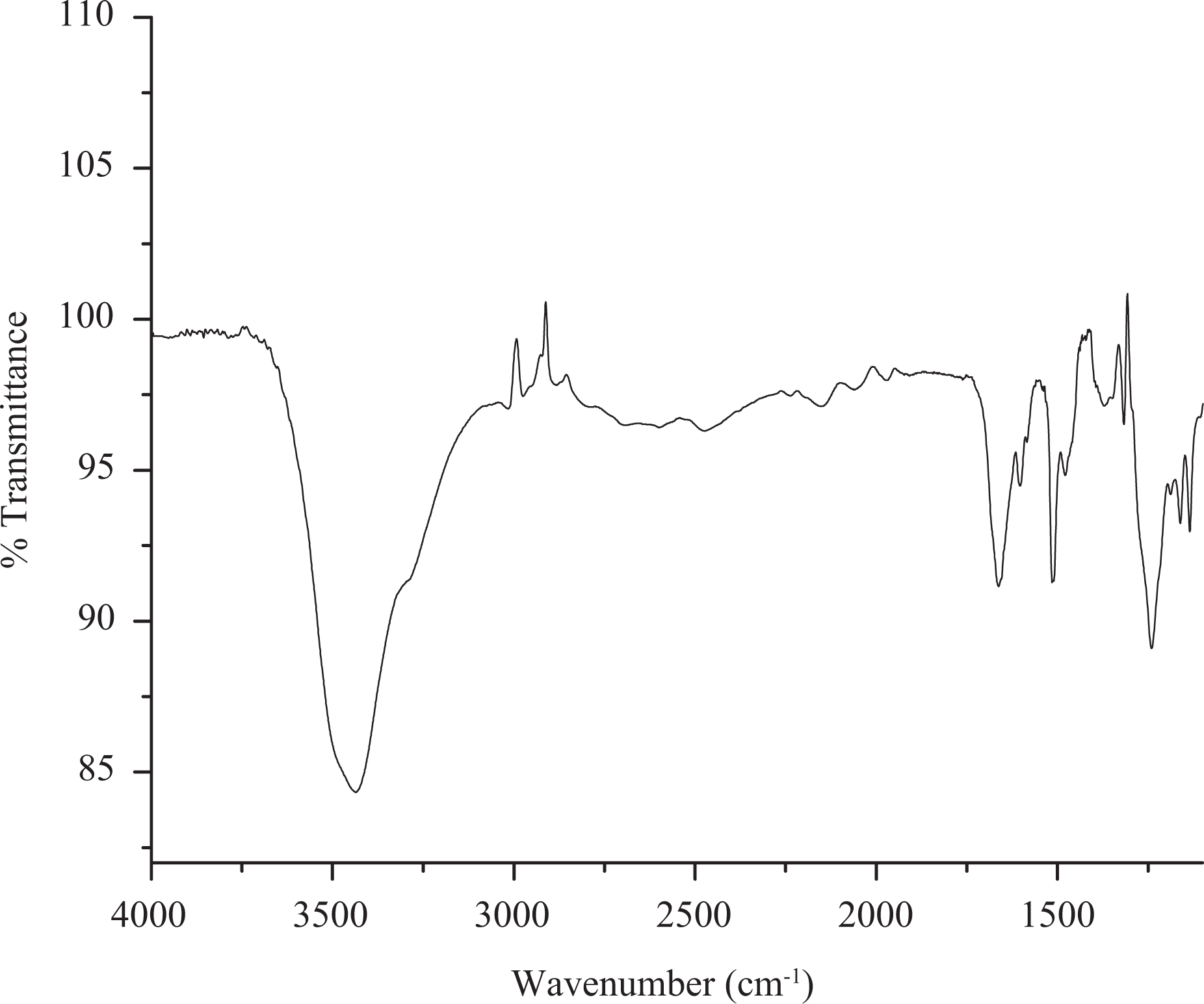
FTIR spectrum of polymer P1. FTIR: Fourier-transform infrared spectroscopy.
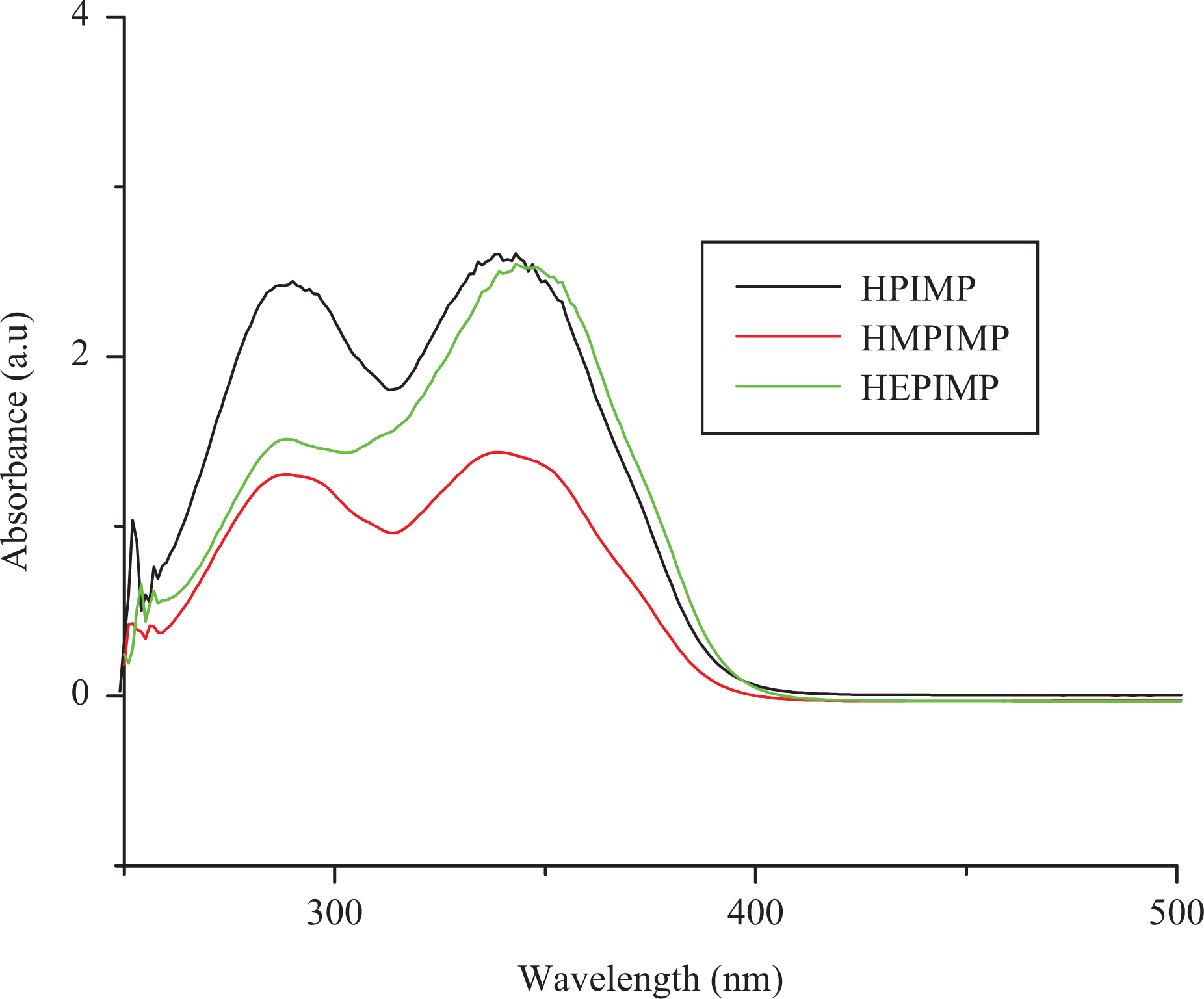
UV–Vis spectra of monomers. UV–Vis: ultraviolet-visible.
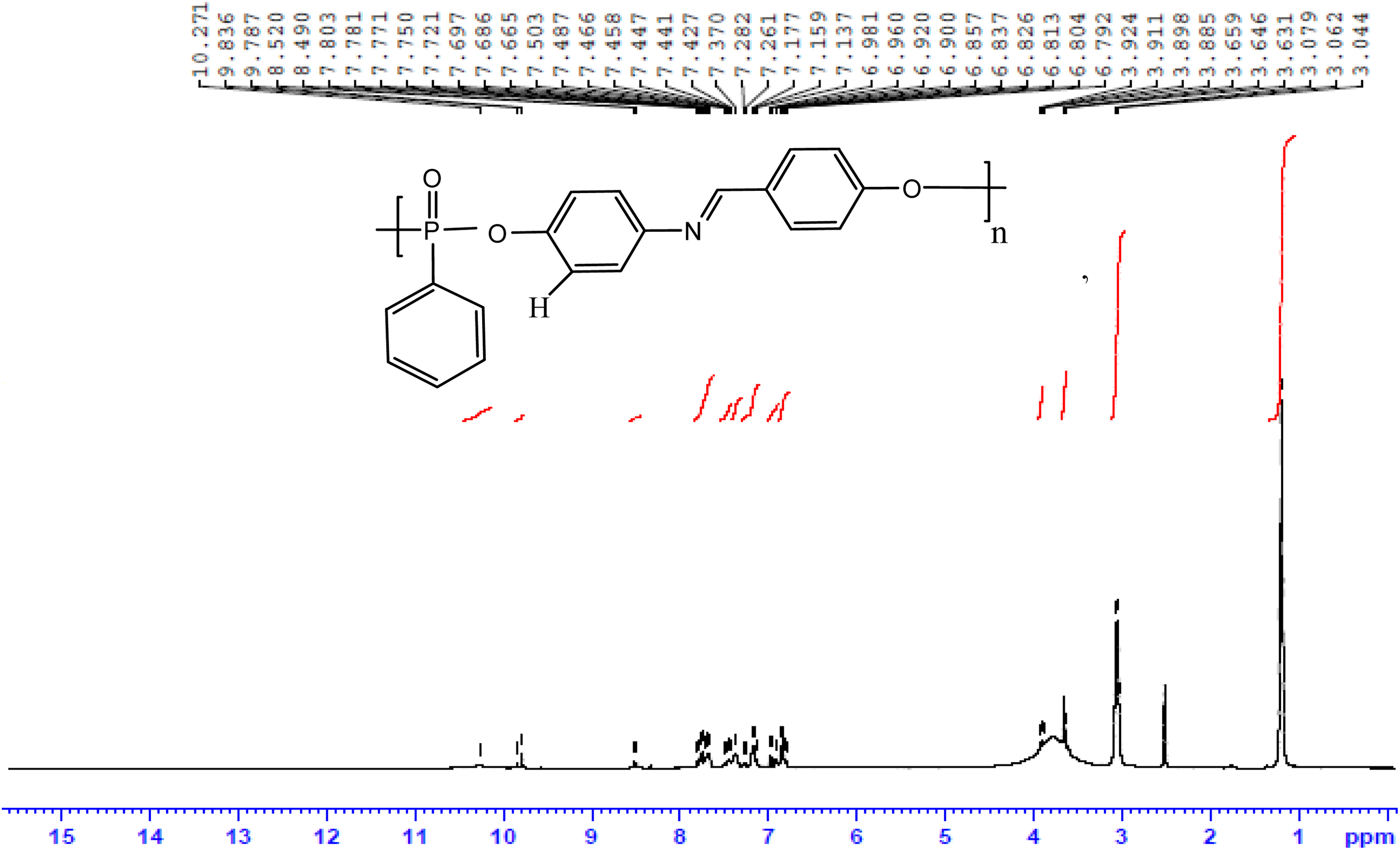
1H NMR spectrum of polymer P1.
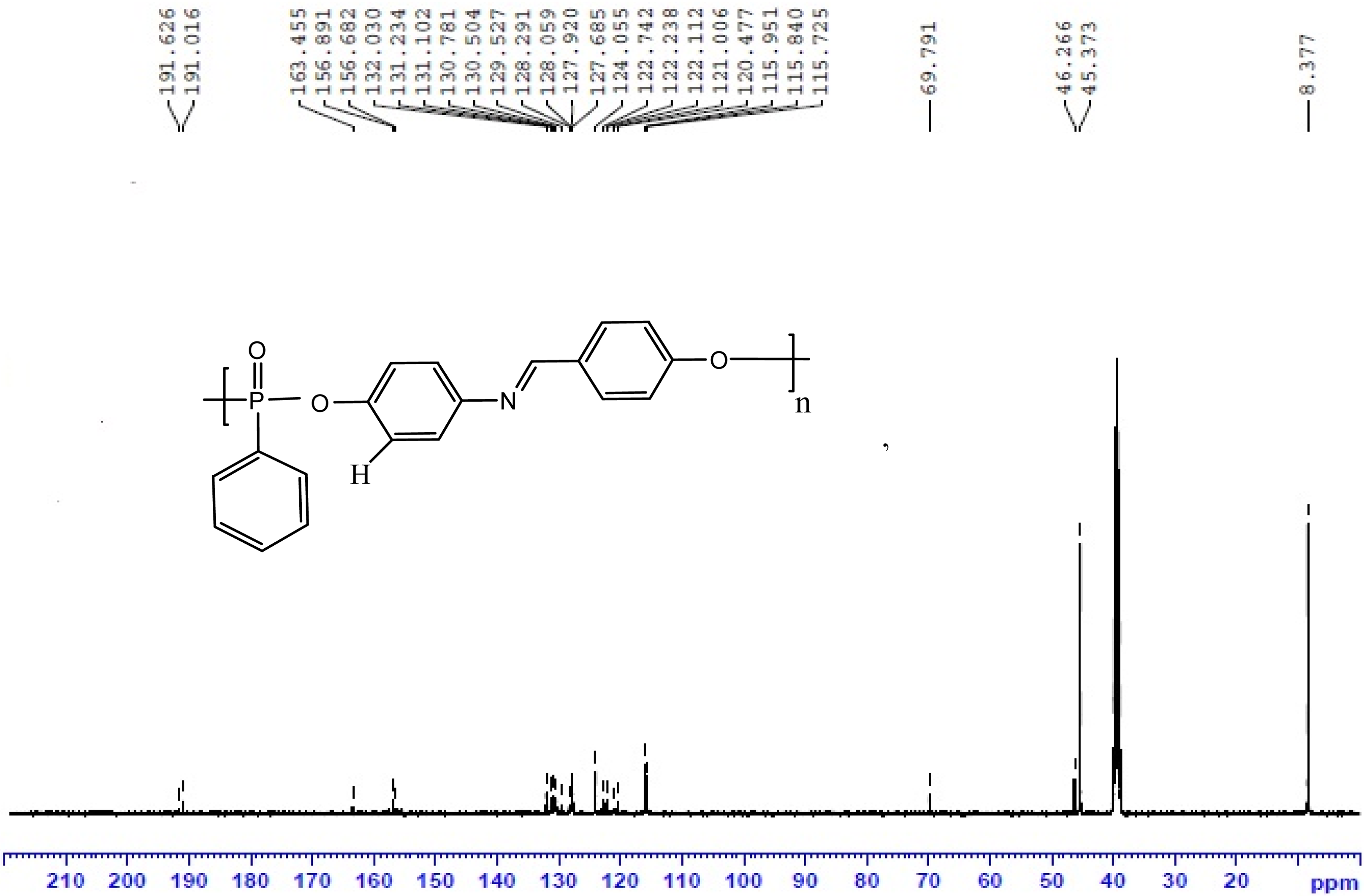
13C NMR spectrum of polymer P1.
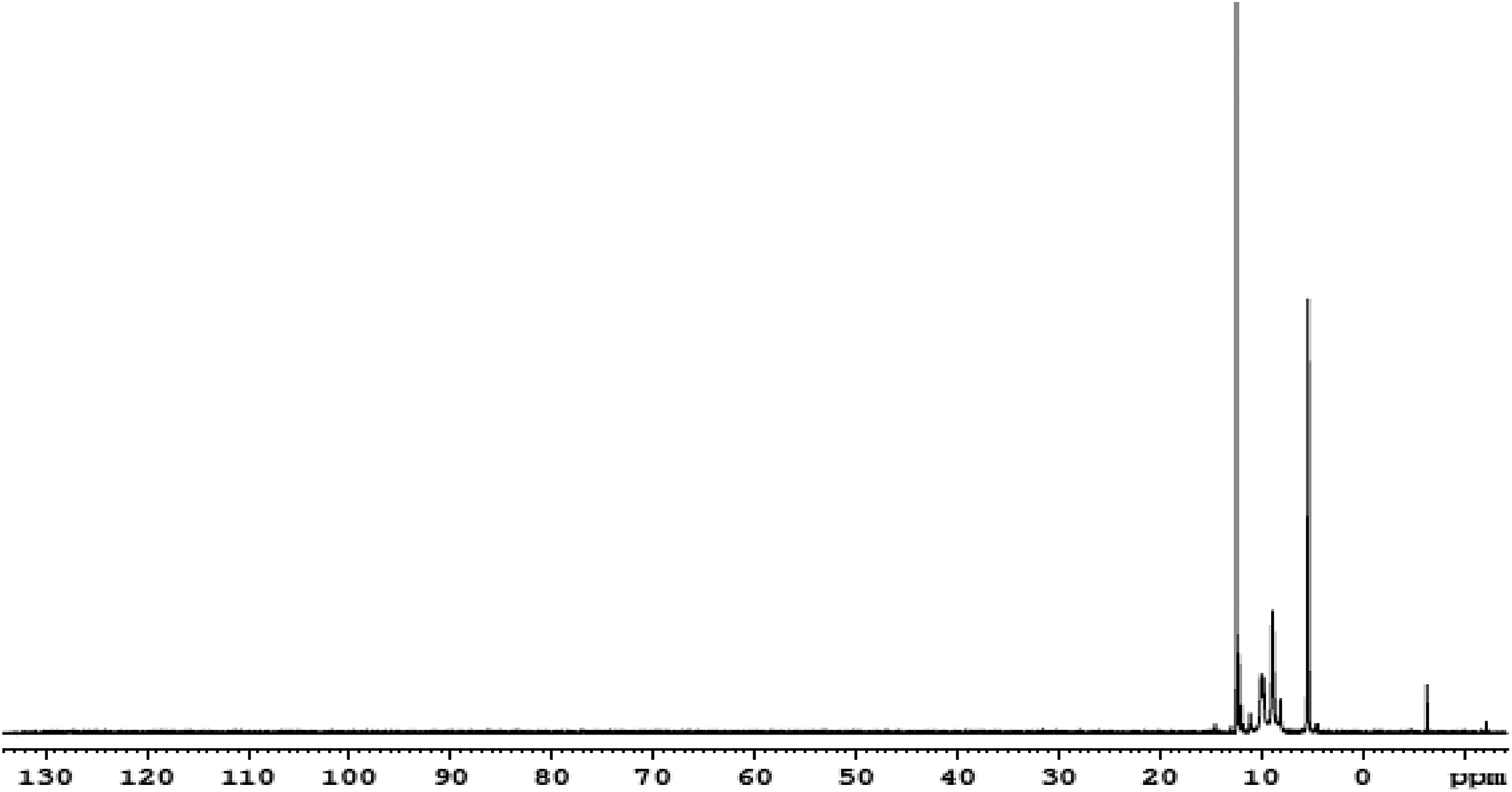
31P NMR spectrum of polymer P1.
Photophysical properties
UV–visible spectra of the monomers in DMSO are shown in Figure 4. The absorption bands of the monomers were observed at 292, 281 and 289 nm suggesting the π→ π* transition of the benzene ring and the absorption bands at 338, 334 and 346 nm were assigned to the π → π* transition of the azomethine moiety. The band due to π → π* transition at 330–360 nm is more sensitive to structural effects and the bisphenols without substituents exhibited bathochromic shift in the absorption band. The optical band gap (E g) was calculated using the equation E g = 1242/λonset, 38 where λonset is the onset wavelength, which can be determined by the intersection of two tangents. The optical band gap values of monomers are found to be 3.67, 3.71 and 3.58 eV for HPIMP, HMPIMP and HEPIMP, respectively.
Thermal properties
Thermal properties of the synthesized polyphosphonates were studied using TGA under nitrogen atmosphere. The TGA traces of all the polymers are shown in Figure 8. The temperatures corresponding to 10, 20, 30, 40 and 50% weight loss were determined from the thermograms and are summarized in Table 3. The results revealed that the aromatic polyphosphonates exhibit single-step degradation. It can be seen that the polymer P1 is stable up to 189°C and 10% weight loss occurred at around 195°C. The polymer P2 is stable up to 176°C and 10% weight loss occurred at around 181°C. The polymer P3 is stable up to 61°C and 10% weight loss occurred at around 68°C. 10% weight loss between 68°C and 189°C is attributed to the loss of moisture, adsorbed solvent or monomer. 39,40 The polymer P1 derived from HPIMP possess higher thermal stability than the polymers P2 and P3 derived from HMPIMP and HEPIMP, respectively. The relatively lower thermal stability of the polymers P2 and P3 may be due to the presence of –OCH3 and –OCH2CH3 substituents in the aromatic ring of the polymer chain. 41 The polymers P1 and P2 produced higher char yield than polymer P3. In polymer P2, the presence of methoxy group needs higher energy for bond cleavage than P3 in which ethoxy group is present. The increase in carbon chain length increases flexibility and decreases the thermal stability. When a phosphorous-containing polymer is heated, cross linking carbonization occurs during the decomposition process. The high char yield limits the production of combustible gases, decreasing the exothermicity of the pyrolysis reactions of the polymers and inhibits the thermal conductivity of the burning materials, thus increasing the flame retardancy of the polymers. 42
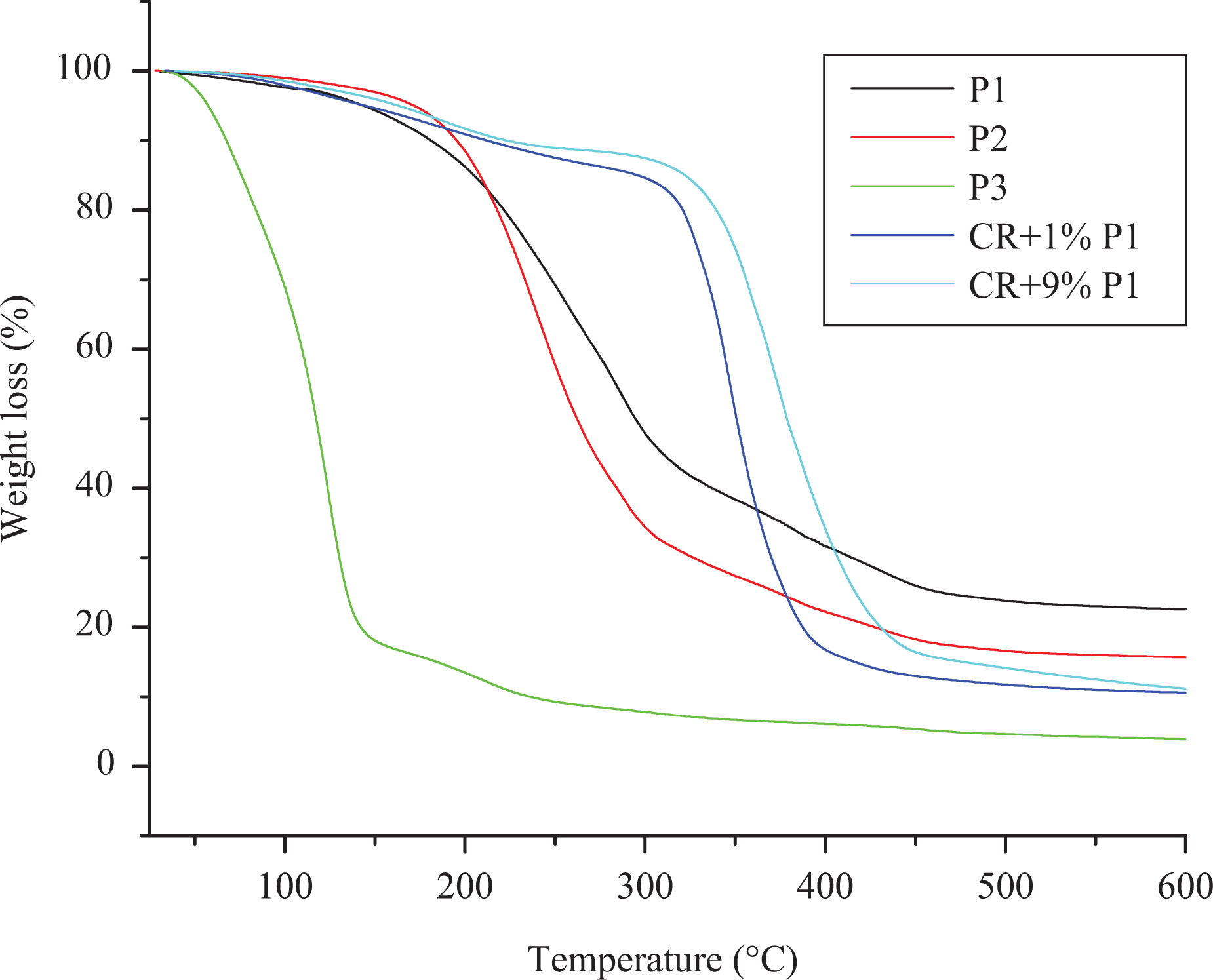
TGA traces of the polyphosphonates. TGA: thermogravimetric analysis.
IR and NMR spectral data of monomers.
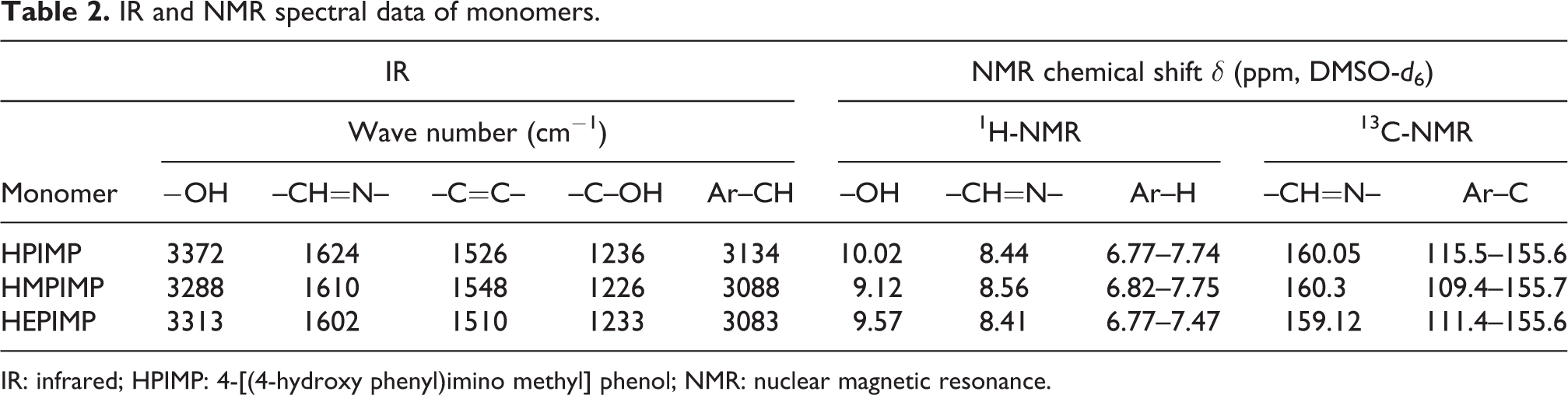
IR: infrared; HPIMP: 4-[(4-hydroxy phenyl)imino methyl] phenol; NMR: nuclear magnetic resonance.
TGA data of the polyphosphonates.

LOI: limiting oxygen index; CR: commercial epoxy resin; TGA: thermogravimetric analysis.
aDetermined by TGA.
bDetermined with Van Krevelen’s equation.
The theoretical LOI values for all of the polymers were determined using the Van Krevelen equation 43 and was in the range of 19–27. According to Van Krevelen, polymers having LOI above the threshold value of 26 possess self-extinguishing property. 44 The obtained LOI values indicated that the polymer P1 has self-extinguishing property. Thermal properties of blended thermoset specimen samples (CR + 1% P1) and (CR + 9% P1) have been studied. The sample (CR + 9% P1) shows higher char yield than the sample (CR + 1% P1). This may be due to higher thermal stability of commercial resin than the synthesized phosphorous-containing polymer. In DSC trace (Figure 9), the appearance of multiple peaks at around 167–216°C indicated the melting of microcrystalline regions present in the hard microdomains. 30 Crystalline transition temperature was observed only for P1 and P2 but not for P3 due to the amorphous nature of polymer. The decrease in crystallinity for polymers P2 and P3 compared to polymer P1 attributed to the flexibility of the polymer chain due to the incorporation of alkoxy linkages in the phenyl groups.
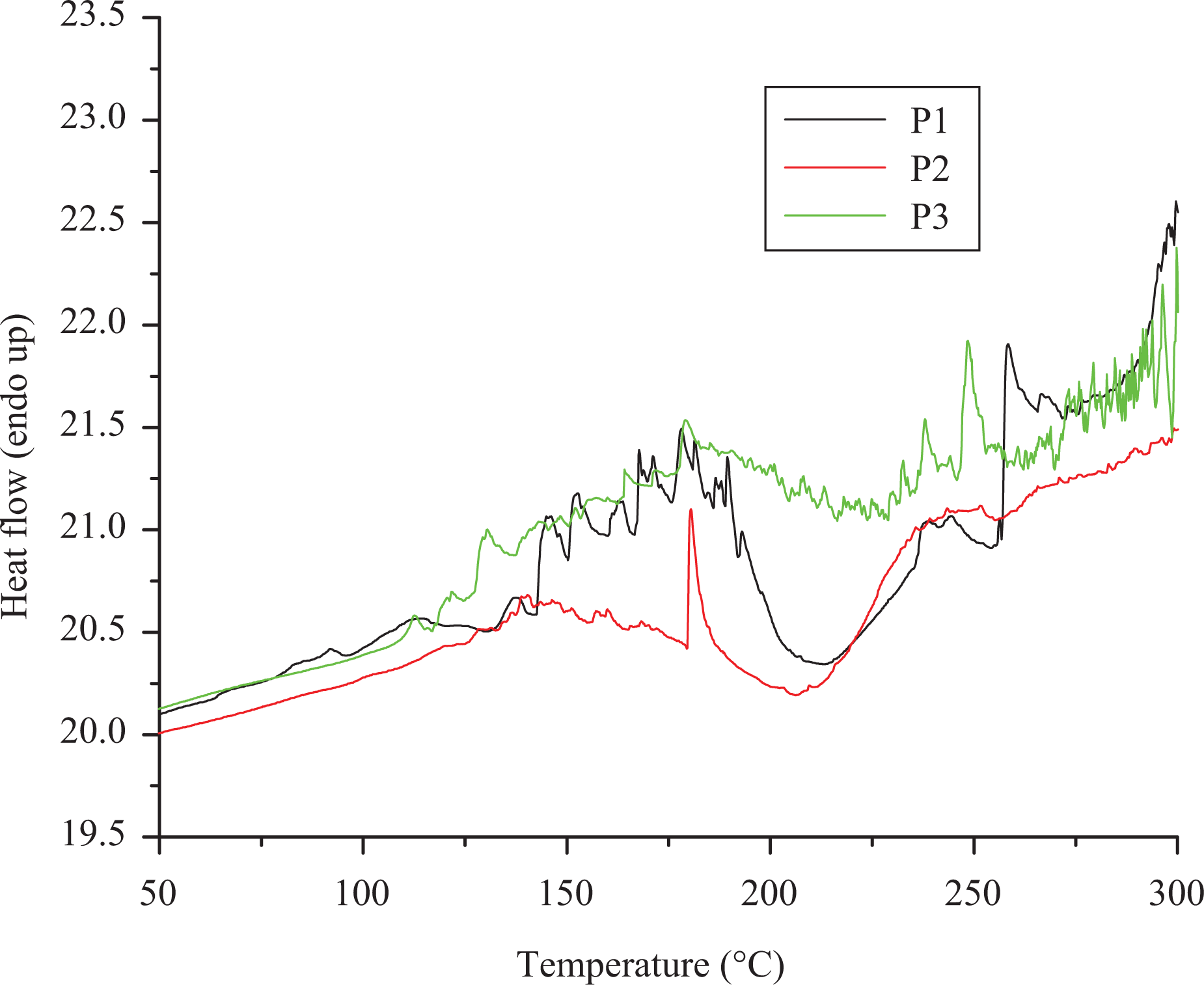
DSC traces of the polyphosphonates. DSC: differential scanning calorimetry.
Kinetic parameters
Kinetic parameters can be correlated with the thermal stability of the polymeric material and it can be used for the prediction of the degradation behaviour of the material in various conditions. In the present investigation, activation energy (E a) for degradation of phosphorous-containing polyesters was determined using Broido and Horowitz–Metzger methods. Broido’s method gives a straight line for the plot of ln [−ln (1 − α)] versus 1/T. In Horowitz–Metzger method, a plot of ln [ln (1/(1 − α))] versus θ gives an excellent approximation to a straight line. From the slope of the straight line, E a was calculated. Representative plot of Broido and Horowitz–Metzger methods are shown in Figures 10 and 11. The regression analysis gives the slopes, constants and concurrency value (R 2) for degradation process. The linear plot with R 2 closer to one was chosen for all methods. The R 2 values and calculated E a values are summarized in Table 4. Among the polymers P1 and P2, P2 has higher E a than the polymer P1. This may be attributed to the presence of methoxy group which need higher energy for Ar–O–C bond cleavage. 45 Among the blended epoxy thermosets, (CR + 1% P1) shows higher E a than (CR + 9% P1). This observation reveals that the incorporation of higher amount of synthetic polymer into thermoset leads to higher plasticization, which reduces the thermal stability.
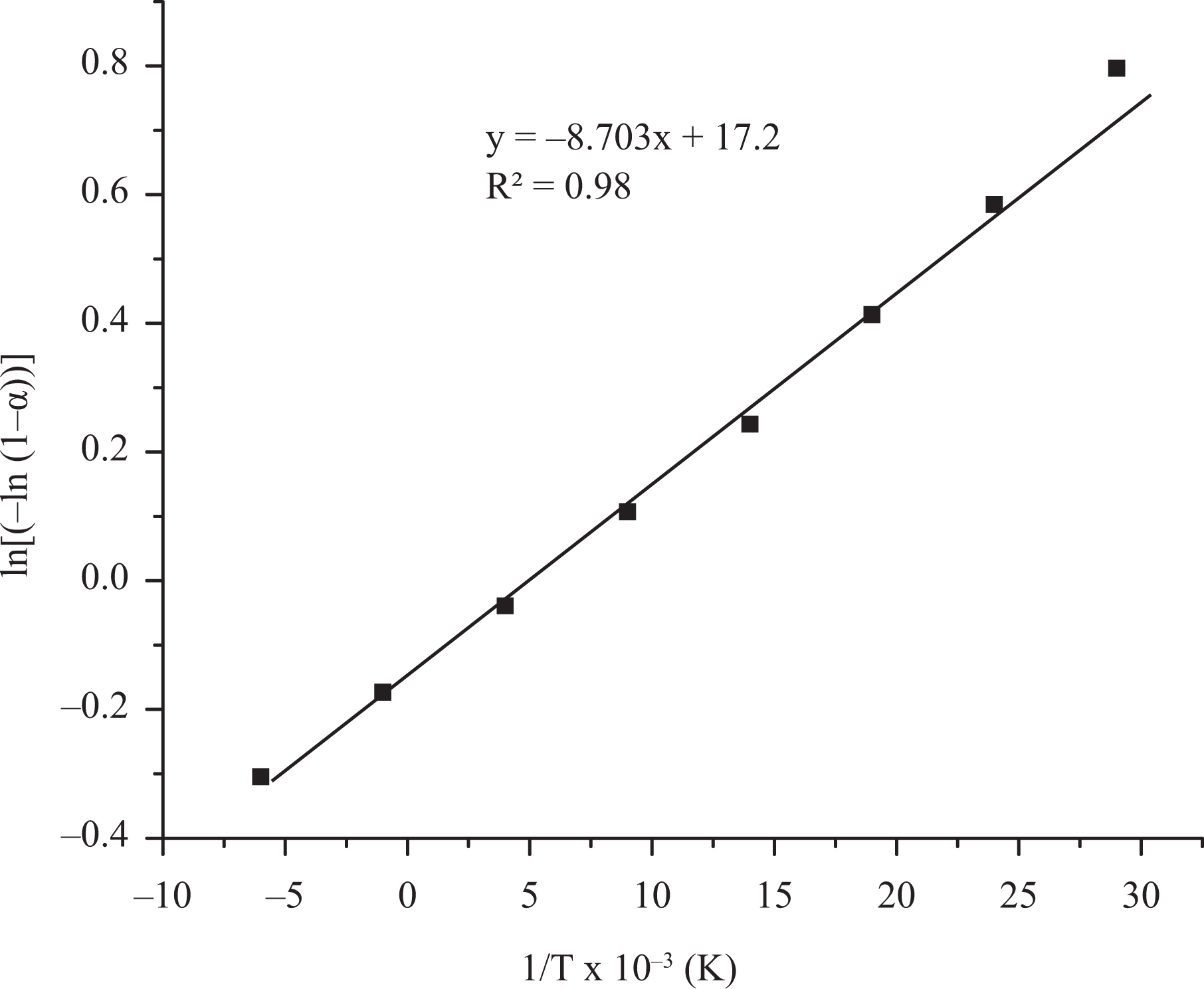
Broido model of polymer P1.
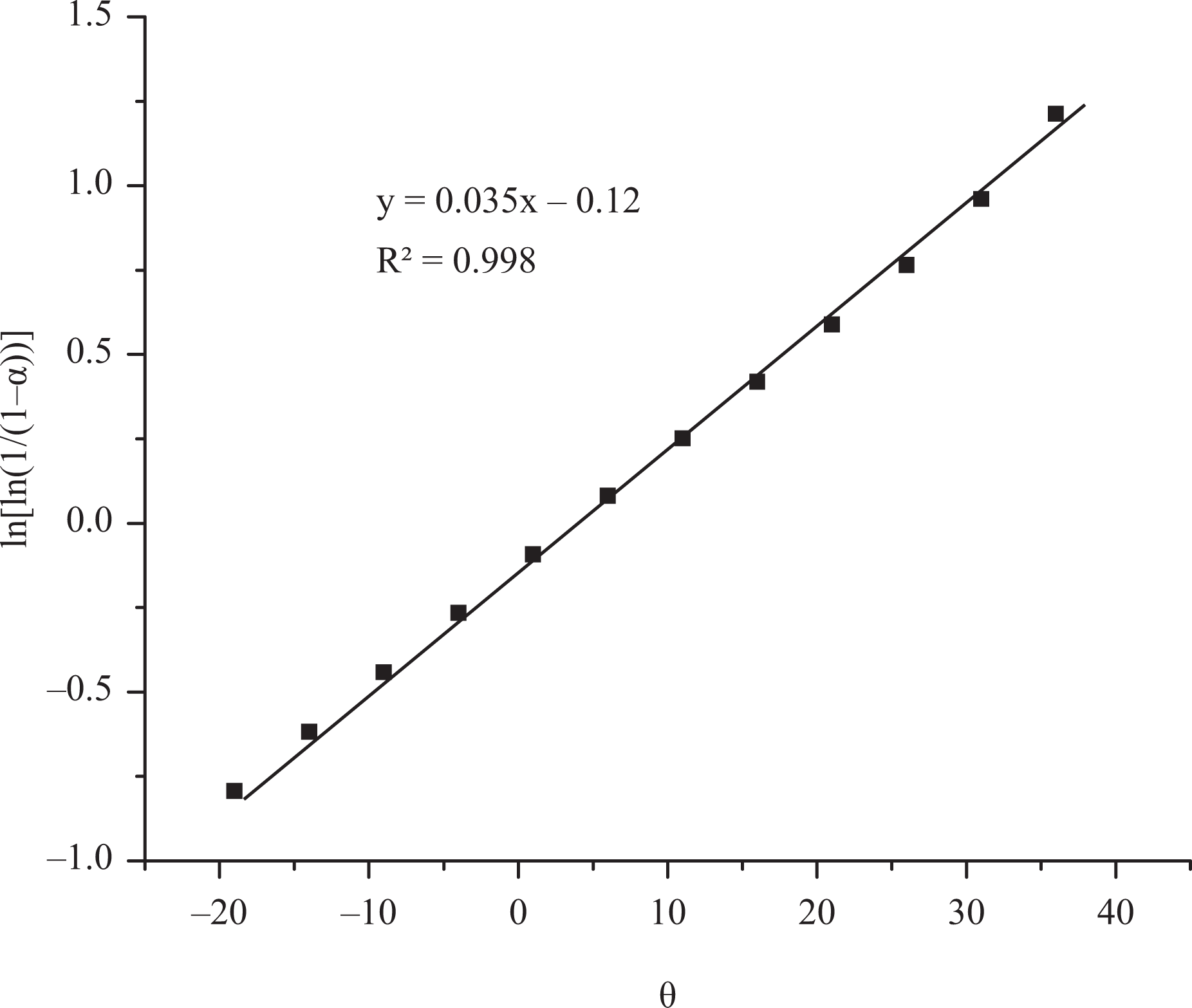
Horowitz–Metzger model of polymer P2.
E a of decomposition and R 2 of polyphosphonates.
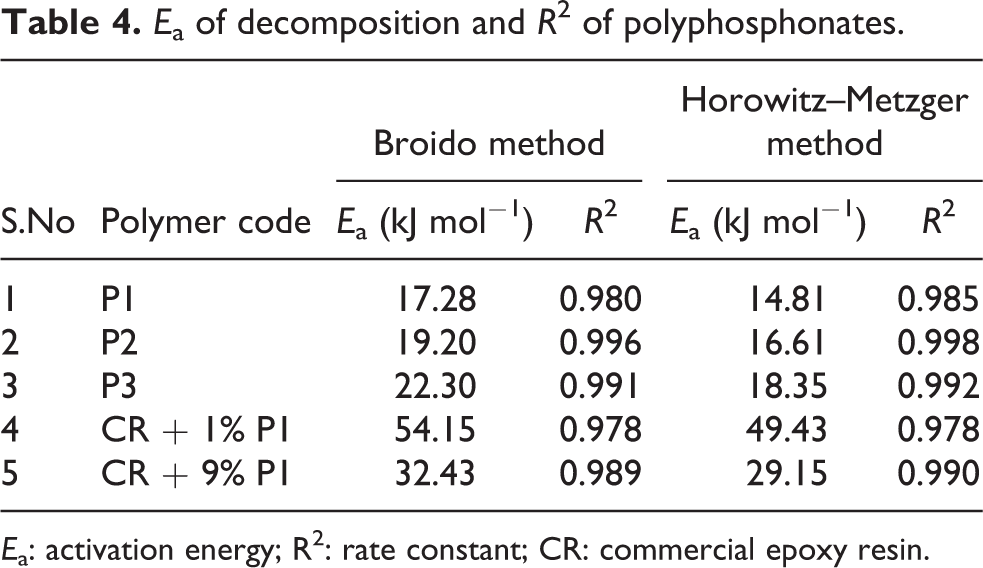
E a: activation energy; R2: rate constant; CR: commercial epoxy resin.
Tensile properties
The polymer samples were subjected to tensile testing and the results are summarized in Table 6. The tensile strength and percentage elongation at break were in the range of 5–36 MPa and 5–18%. The results showed that the tensile strength of specimen (T) is higher than the other specimens incorporated with synthesized polyphosphonate. This may be attributed to the introduction of flexible group like phosphonate, which reduces the cross-linking and intermolecular interaction, thereby increasing the elongation and decreasing the tensile strength. The data also revealed that the specimen TPA has higher tensile strength than the specimen TPE. This may be associated with the increase in percentage of polyphosphonate which decreases the packing density of the molecular chain due to strong intermolecular interactions between polymer chain. These interactions decreased the tensile strength and increased the percentage of elongation to break.
Flame retardancy
The results of UL-94 vertical burning test and LOI tests of the prepared specimen for flame-retardant property of the synthesized azomethine polyphosphonates are shown in Table 5. The result reveals that the addition of polyphosphonates to epoxy resin increases the LOI value as well as increases the rating in UL-94 test. Furthermore, the increase in phosphorous content increases the LOI value. This may be due to high char formation by the phosphorous-containing additives that hinder the diffusion of oxygen to the polymer. In this study, phosphorous acted as a flame retardant element to dilute the concentration of flammable gases produced during the combustion process and also decreased the heat release from the polymer samples. The flame-retardant character of the synthesized polyphosphonate was compared with commercial poly phosphonates of FRX polymers. Commercial polyphosphonate, Nofia HM 5000, showed V0 category in UL-94 test, whereas synthesized polyphosphonates showed V1 category in UL-94 test.
Flame retardancy of polyphosphonates.
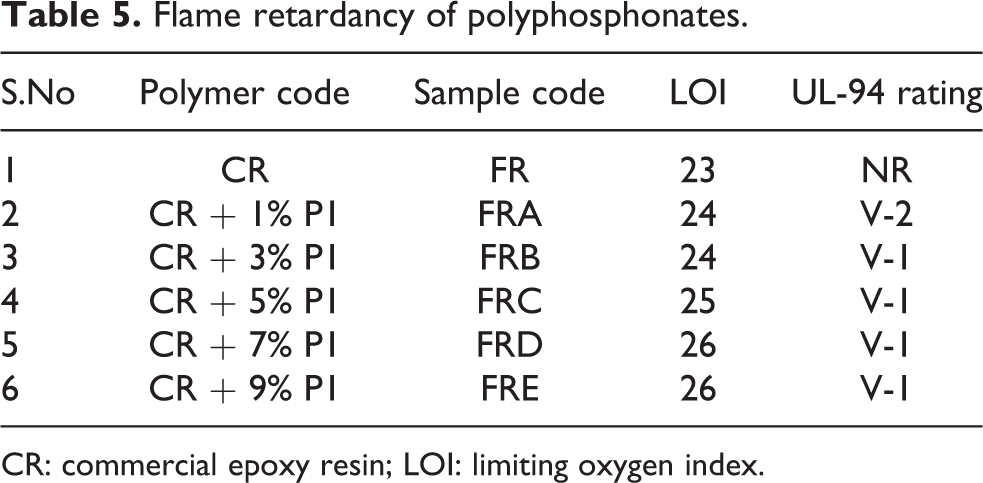
CR: commercial epoxy resin; LOI: limiting oxygen index.
Tensile properties of the polyphosphonates.
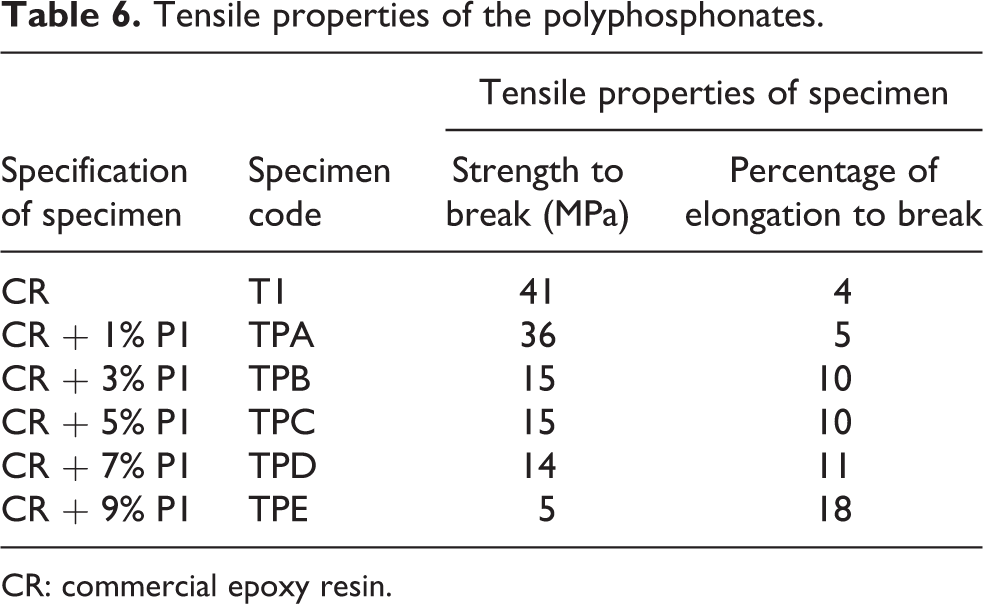
CR: commercial epoxy resin.
Conclusions
Azomethine polyphosphonates were synthesized from various azomethine bisphenols and phenylphosphonic dichloride by solution polycondensation technique using triethylamine as catalyst. The formation of polymers were confirmed using FTIR, 1H-, 13C- and 31P-NMR spectroscopic techniques. The LOI value of polymer P1 is higher than the threshold value of about 26 and thereby they are expected to have self-extinguishing properties. In DSC results, the presence of melting peaks at about 216°C indicated that the synthesized polymers are crystalline in nature. The kinetic parameters of thermal degradation of the polyphosphonates have been analysed by Broido and Horowitz–Metzger methods. The LOI and tensile strength results indicated that the synthesized polyphosphonates have the potential to be used as a flame-retardant plasticizer.
Footnotes
Acknowledgment
The authors would like to thank the Principal and Management of PSG College of Technology, Coimbatore, Tamil Nadu, India, for providing the necessary facilities, the Indian Institute of Technology (Madras), Chennai, Tamil Nadu, India, for nuclear magnetic resonance (NMR) spectral studies and the Sophisticated Test and Instrumentation Centre, Cochin, Kerala, India, for elemental analysis.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.