
Abstract
Novel high-performance polyacrylonitrile (PAN)-based pre-oxidized fibers (i.e. OPFHA-MEA-L) with improved thermal stability and flame-retardant and mechanical properties were designed and made from the pristine PAN fibers through chemical pretreatment with hydroxylamine hydrochloride (HA) and monoethanolamine (MEA) aqueous solutions, then coated with chitosan (CS) and sodium tripolyphosphate (STPP) via layer-by-layer (LbL) assembly, and finally followed by stabilization in the air. The morphological structure, flammability, and thermal and mechanical properties of fabricated OPFs were systemically investigated. The results indicated that the PAN fibers after chemical pretreatment with HA and MEA had a large amount of hydrophilic groups. It would facilitate the increase of pre-oxidation degree for PAN fibers during stabilization and the deposition of positively and negatively charged CS-STPP flame-retardant coating. The fabricated OPFs (i.e. OPFHA-MEA-10) demonstrated superior comprehensive properties with charred residue of about 68.2%, breaking strength of about 295.1 N, breaking elongation of 12.6%, and limiting oxygen index value of about 41.5%, respectively, contributing to the improved thermal stability and flame-retardant and mechanical properties. It is envisioned that this innovative type of high-performance OPFs could be utilized for potential applications as flame retardant and in high temperature filtration.
Keywords
Introduction
Acrylic fibers, the academic name of polyacrylonitrile (PAN) fibers, were based upon acrylonitrile as the main monomer (content higher than 85%) and a small amount of other monomers. 1,2 It had a wide range of uses, abundant raw materials, and rapid development, and it was now one of three major synthetic fibers. 3 –5 However, the PAN fibers were flammable with low limiting oxygen index (LOI) of about 18%. When it contacted a high-temperature and/or heat source, it was easy to burn and thus cause fire. This not only caused great damages to people’s lives and properties but also seriously harmed our environment. 6,7 Meanwhile, due to the limitations of PAN itself and the production process, the research and development of flame-retardant PAN fibers lagged far behind that of polyester, polypropylene, and viscose. Therefore, under the background of increasingly fierce market competition, it was of great practical significance to develop a viable PAN flame-retardant process and realize the production of flame-retardant PAN fibers as soon as possible.
The PAN-based pre-oxidized fibers (OPFs) were the novel flame-retardant fibers, which were made from the pristine PAN fibers through stabilization in a muffle furnace with air atmosphere, certain tension, and temperature (e.g. about 200–300°C). 8 –10 Through a pre-oxidation treatment for a certain period of time, the cyano (–C≡N) groups underwent cyclization, dehydrogenation, and oxidation reactions to form heat-resistant ladder and/or conjugated structures. 11 –13 However, due to the effects of thermal oxidation process, the oxidation efficiency was still low and the product’s price was high. At the same time, a large amount of heat generated during pre-oxidation process led to poor and/or low elongation for fibers, which had become one of the important factors affecting the structure and performance of the OPF. 14,15
The chemical modification referred to a method of changing the fibers’ chemical compositions through various techniques, including a crosslinking reaction of a –C≡N group, a cyclization reaction, and a grafting flame-retardant element and/or group. 16 –19 Recently, the PAN fibers were chemically pretreated with hydroxylamine or anthraquinone by utilizing high activity of –C≡N groups on the PAN macromolecular chains. For example, Ren et al. grafted acrylic acid (AA) onto PAN fabrics (PAN-g-PAA). Then, the PAN-g-PAA was modified sequentially by acylation, ammonization, and phosphorylation to obtain acylated PAN-g-PAA, ammoniated Cl-PAN-g-PAA, and fire-retardant PAN fabrics, respectively. The LOI value of the fire-retardant fabrics could reach up to 28.1%. 20 Zhang et al. modified PAN fabrics with hydroxylamine hydrochloride (HA) to prepare amidoxime PAN fabrics (A-PAN) followed by phosphorylation with phosphoric acid (PA) to obtain flame-retardant PAN fabrics (P-A-PAN). The fabricated P-A-PAN demonstrated excellent thermal stability and high LOI value of 34.1%. 21
Moreover, the environmental protection and green materials had also drawn continuous attentions for decades. 22,23 It not only required to achieve flame-retardant PAN fibers but also demanded halogen-free, low-smoke, and low-toxicity flame retardant. 24 Therefore, the intumescent flame retardant became the best strategy for halogen-free flame-retardant technology. It was composed of a gas source, an acid source, and a carbon source. 25 –27 The gas source was thermally decomposed by a nitrogen-containing compound to form a hard gas such as N2 and NH3. These released gases could dilute the air around the surface of the heated materials. The acid source was a phosphorus-containing compound, which was thermally oxidized to form metaphosphoric acid, PA, and polyphosphoric acid. It would participate in the dehydration and carbonization reactions of the carbon source. The carbon source was rapidly degraded and carbonized to form a dense oxide layer when the material was heated. Finally, they formed a nonvolatile, dense, and stable carbonaceous and phosphocarbonaceous residue, which covered the surface of the burning materials and acted as functions of heat-insulating and oxygen barrier. Edwards et al. synthesized two novel phosphoramidate flame-retardant monomers, and they were applied in cotton textiles by using atmospheric pressure dielectric barrier discharge plasma. The results demonstrated the effectiveness of the grafted phosphorus-containing polymer at promoting char formation. 28 Fang et al. designed polyhexamethylene guanidine phosphate–ammonium polyphosphate (PHMGP-APP) assembly coating on cotton fabric to achieve enhanced flame retardant and antimicrobial treatment by layer-by-layer (LbL) technology. The coating performed perfect flame-retardant property of cotton owing to the synergistic effect of PHMGP with APP by facilitating the formation of char. 29
Despite the reported investigation on flame-retardant cotton fabric through LbL assembly technology, 26,29 to the best of our knowledge, the strategy combining chemical pretreatment, LbL assembly, and stabilization techniques for the investigation of flame-retardant PAN fibers has not been reported. The chemical pretreatment will promote chemical reactions between cyano (–C≡N) groups of PAN fibers and chemical reagents (HA and monoethanolamine (MEA)), make the PAN fibers from lipophilic to hydrophilic, and thus provide the convenience for the following LbL assembly. Meanwhile, it will also facilitate the increases of –C≡N conversion rate and promotes the occurrences of cyclization reactions for PAN fibers during stabilization process. The LbL assembly technique, by alternating the deposition of positively and negatively charged substances on substrates (e.g. textile fabric), is one of the most promising methods capable of enhancing flame retardancy. The stabilization promotes the constructions of heat-resistant ladder and/or conjugated structures for PAN fibers via cyclization, dehydrogenation, and oxidation reactions of –C≡N groups. Herein, we design and fabricate novel high-performance OPFs with excellent thermal stability and flame-retardant and mechanical properties. The OPFs are made from pristine PAN fibers through chemical pretreatment with mixed reagents of HA and MEA, then coated with chitosan (CS)–sodium tripolyphosphate (STPP) system via LbL assembly as an intumescent flame-retardant system, and followed by stabilization in air. The CS-STPP flame-retardant coating has nontoxic, biodegradable, and reproducible properties, and also decreases thermal damage for PAN fibers during stabilization. The method has the advantages of simple operation, environment-friendly, water-based system and the use of non-halogen chemicals. The fabricated OPFs are believed to have promising and potential applications in flame retardant and high temperature filtration.
Experimental
Materials
Commercial PAN fibers were kindly provided by Jilin Chemical Industrial Company Ltd (China). HA, MEA, CS, STPP, anhydrous sodium carbonate (Na2CO3), acetic acid (HAc), sodium hydroxide (NaOH), and alcohol were purchased from Sinopharm Chemical Reagent Co., Ltd (China) and were analytically pure. The chemicals/materials were used as received without further purification.
Preparation of PANHA-MEA fibers by chemical pretreatment
The pristine PAN fibers were firstly washed with alcohol to remove oil traces and impurities on the fibers’ surface. Meanwhile, a certain concentration of HA (40 g L−1) and MEA (30 mg L−1) solution was configured, respectively. Then, the PAN fibers were pretreated in the HA aqueous solution at 80°C for 2 h (solution pH value was adjusted to 5 with anhydrous Na2CO3), washed with deionized water and dried in an oven at 50°C. Subsequently, the fibers were added into the above MEA aqueous solution. The reaction mixtures were refluxed at 90°C under stirring for 2 h. The reaction products were cooled to room temperature, washed with deionized water, and then dried in an oven at 70°C to obtain PANHA-MEA fibers. The possible chemical reactions that happened during pretreatment are presented in Figure 1.
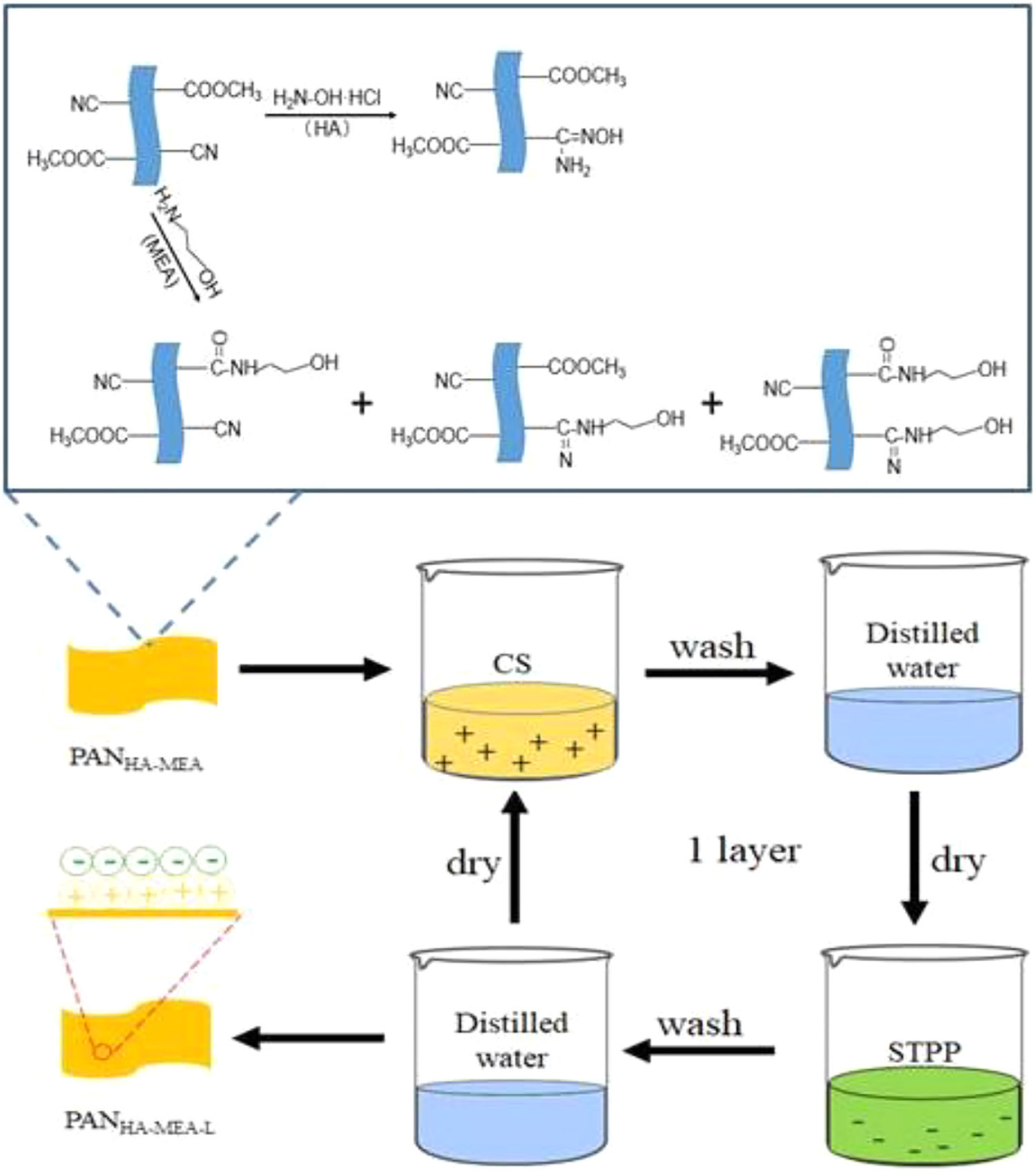
Schematic illustration of the fabrication process for PANHA-MEA-L fibers. PAN: polyacrylonitrile; MEA: monoethanolamine; HA: hydroxylamine hydrochloride.
Fabrication of PANHA-MEA-L fibers coated with CS-STPP by LbL assembly
CS solution (1.0 wt%) was prepared by adjusting the solution pH value to 5 with 1 M HAc and then was magnetically stirred until the CS powders were completely dissolved. Meanwhile, 2.0 wt% STPP solution was also prepared by dissolving STPP into deionized water, and the solution pH value was adjusted to 4 with 1 M NaOH aqueous solution.
The above prepared PANHA-MEA fibers were firstly immersed in CS solution to obtain a layer polycation and dried in an oven at 90°C. Then, the fibers were soaked into STPP solution to obtain a layer polyanion which could pair with CS through electrostatic forces, and dried in an oven at 90°C. Subsequently, PANHA-MEA fibers were alternately dipped into positive (CS) and negative (STPP) solutions with the procedure, as shown in Figure 1. The initial dip was 15 min and subsequent dips were controlled for 5 min. After the desired number of bilayers was deposited, the fibers were dried at 50°C in an oven and were defined as PANHA-MEA-L fibers (i.e. PANHA-MEA-5, PANHA-MEA-10, PANHA-MEA-15, and PANHA-MEA-20), where L represented the numbers of assembly bilayers.
Fabrication of OPFs
The PANHA-MEA-L fibers were placed in a muffle furnace with a certain tension and subjected to elevated temperature stabilization in the air. The stabilization starting temperature was 170°C, followed by raising the temperature to 240°C with the heating rate of 1°C min−1 and maintaining the temperature for 10 min. Thereafter, the stabilized fibers were naturally cooled to room temperature for making the OPF (i.e. OPFHA-MEA-L). Meanwhile, the OPF and OPFHA-MEA were also obtained under the same experimental conditions.
Characterization
The contact angle measurement was taken at six different locations per sample with 5 µL deionized water using a surface tension dynamic contact angle measuring instrument (DCAT-21, Dataphysics Instruments Co., Germany). Fourier transfer infrared (FTIR) spectra were acquired from a Nicolet MAGNA-IR 750 (American Thermo Fisher Scientific Co., Ltd.) spectrometer in the wavenumber range of 400–4000 cm−1 with a resolution of 4 cm−1. During the preparation of FTIR specimen, a small amount of a testing sample was ground together with pre-dried KBr in a mortar into a uniform mixture; subsequently, the mixture was pressed into a standard thin disk. The amount of each sample was approximately the same for FTIR analysis. The morphologies of OPF, OPFHA-MEA, and OPFHA-MEA-L were characterized using scanning electron microscopy (SEM; SU1510, Hitachi Co., Ltd., Japan) with an operating voltage of 20 kV. Prior to SEM examination, all specimens were sputter-coated with gold to avoid charge accumulations. Chemical compositions of samples were simultaneously carried out by energy dispersive spectrometry (EDS) during the SEM investigation. The crystal structures of samples were tested on X-ray diffractometer (XRD; Bruker D8, 40 kV, 40 mA, Germany) with Cu-Kα radiation (wavelength λ = 1.54 Å) at a scanning speed of 0.02° s−1 with a 2θ range of 5–40°.
The mechanical property was measured using an electronic single yarn strength tester (YG(B) 026 D-250, Wenzhou Darong Textile Standard Instrument Co., Ltd., China). The samples were conducted at a stretching speed of 20 mm min−1 with a gauge length of 20 mm. For each data point, five samples were tested and the average value was taken. Thermogravimetric analyses (TGA) were carried out using a TA Instrument (American TA Instruments) Q500 thermo-analyzer instrument; the heating rate was set at 10°C min−1, and the TGA curves were recorded from 30°C to 800°C under 25 ml min−1 flow of nitrogen. The amount of each sample for the TGA analyses was approximately 10 mg. LOI measurement was carried out on a JF-3 LOI analyzer (Beijing Xinsheng Excellence Technology Co., Ltd., China), and the samples were made by twisting fibers according to the FZ/T 50017-20 standard.
Result and discussion
Chemical compositions
The surface characteristics of pristine PAN fibers before and after chemical pretreatment were roughly evaluated by water contact angle, as shown in Figure 2. It could be seen from Figure 2(a) that the pristine PAN fibers were highly hydrophobic with water contact angle of about 109.6°. The shape of the droplet was almost spherical on the surface, when water droplet was dropped on the fibers’ surface. However, after chemical pretreatment with mixed reagents of HA and MEA, the water droplet spread on the surface of fibers, and the fibers exhibited notable hydrophilic, as shown in Figure 2(b). This was because a large number of hydrophilic groups (e.g. –OH, –NH, and –NH2) appeared on the fibers after chemical modification, due to chemical reactions between –C≡N groups of PAN fibers and chemical reagents (HA and MEA). This would be in favor of absorbing CS via electrostatic interactions in the following LbL assembly. It was also envisioned that the surface characteristics of fibers would facilitate the assembling of CS-STPP flame-retardant coating.

Contact angles of (a) pristine PAN fibers and (b) PANHA-MEA fibers through chemical pretreatment. PAN: polyacrylonitrile; MEA: monoethanolamine; HA: hydroxylamine hydrochloride.
To confirm the available functional groups on the fibers’ surface, the pristine PAN fibers, modified PAN fibers after chemical pretreatment (i.e. PANHA-MEA), modified PAN fibers through chemical pretreatment followed by LbL assembly (i.e. PANHA-MEA-20), and OPFs (i.e. OPFHA-MEA-20) were characterized by FTIR spectra, as shown in Figure 3. In the spectrum of PAN fibers, the bands located at about 2939 and 2850 cm−1 were assigned to the stretching vibrations of C–H groups. The bands centered at 2243 and 1470 cm−1 were attributed to the stretching vibrations of –C≡N groups and the bending vibrations of methylene (–CH2–) groups, respectively. The band centered at 1700 cm−1 was due to the stretching vibrations of carbonyl (C=O) groups in the comonomers of itaconic acid and methyl acrylate. 21,30 In comparison with FTIR spectrum of the PAN fibers, the characteristic peak of the –C≡N groups at 2243 cm−1 for PANHA-MEA fibers was significantly decreased. The characteristic peak centered at 1700 cm−1 shifted to lower wavenumber and a new and board characteristic peak occurred at about 1647 cm−1, due to the overlaps of the stretching vibrations of C=O and C=N groups. Meanwhile, the occurrences of new characteristic peaks located at about 1593 and 911 cm−1 were assigned to the stretching vibrations of amidoxime (C–NH2) and N–O groups, respectively. The results further confirmed that there were chemical reactions between –C≡N groups of PAN fibers and chemical reagents (HA and MEA), reducing the activation energy required in the pre-oxidation process. In the spectrum of PANHA-MEA-20, it could be clearly seen that the strength of hydroxyl (–OH) characteristic peak at about 3000–3750 cm−1 was remarkably increased after LbL assembly, which was attributed to the fact that the CS macromolecular side chains involved a large amount of –OH groups. But it was difficult to distinguish precisely the characteristic absorption peaks at 1164 and 981 cm−1 of STPP (e.g. P=O, O–P–O, and P–O), due to overlaps of characteristic absorption peaks. 3 After the stabilization, the FTIR spectrum of OPFHA-MEA-20 showed that the characteristic absorption peaks of –C≡N and C–NH2 groups significantly decreased and/or disappeared, indicating most of –C≡N groups, if not all, was converted into ladder and/or conjugated structures.
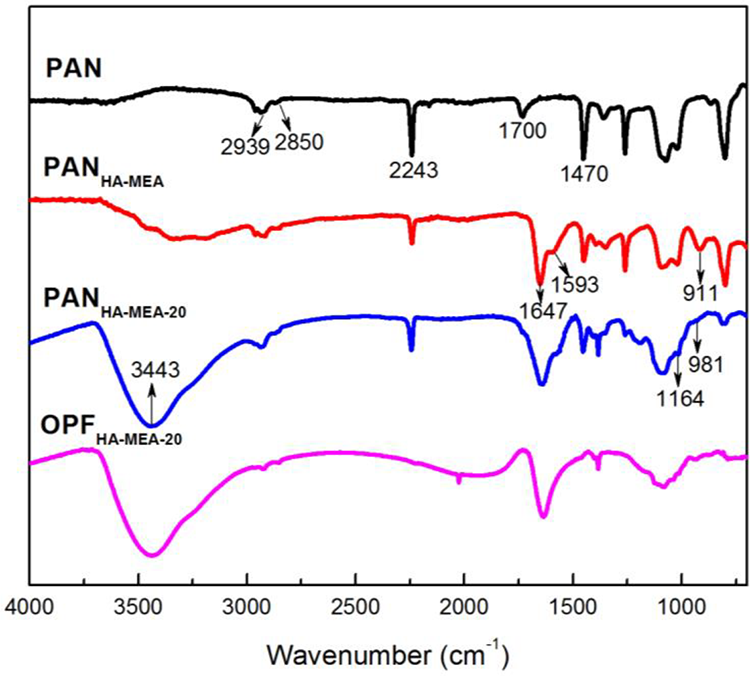
FTIR spectra of PAN, PANHA-MEA, PANHA-MEA-20, and OPFHA-MEA-20. FTIR: Fourier transfer infrared; PAN: polyacrylonitrile; OPF: polyacrylonitrile-based pre-oxidized fiber; MEA: monoethanolamine; HA: hydroxylamine hydrochloride.
In order to prove the successful modification of PAN fibers and the existence of P element, the chemical compositions of the modified PAN fibers through chemical pretreatment followed by LbL assembly (i.e. PANHA-MEA-20) were analyzed by EDS. Figure 4(a) showed that the PANHA-MEA-20 fibers mainly contained C, N, O, and P elements. For further determining element distribution on the fibers’ surface, SEM elemental mapping was also performed for the PANHA-MEA-20 fibers, as shown in Figure 4(b). The result indicated that all the above elements were detected and dispersed homogeneously on the surface of fibers. This demonstrated that the CS-STPP coating was successfully deposited on the PANHA-MEA fibers’ surface through LbL assembly.
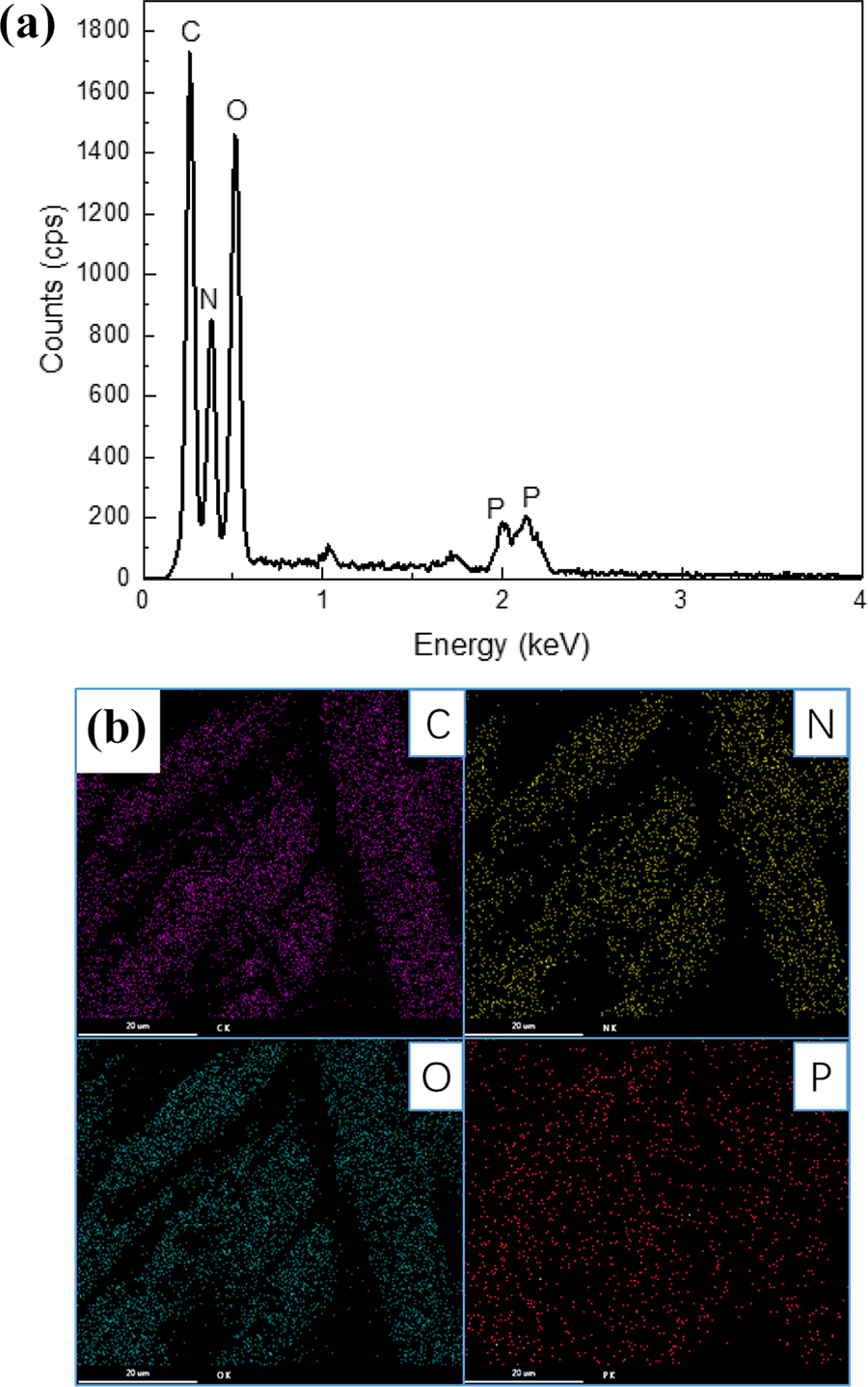
(a) EDS spectrum and (b) elemental mapping of the PANHA-MEA-20. EDS: energy dispersive spectrometry; PAN: polyacrylonitrile; MEA: monoethanolamine; HA: hydroxylamine hydrochloride.
Morphological structure
Figure 5 demonstrated the representative SEM micrographs of pristine PAN fibers and OPFs (i.e. OPF, OPFHA-MEA, and OPFHA-MEA-L). Figure 5(a) showed that the surface of pristine PAN fibers was relatively smooth and slightly covered with cracks and grooves due to the formed micro-defect structures during the manufacturing process. It could be seen from Figure 5(b) that the surface of the OPF formed some superficial cracks. This was because the fibers’ surface not only increased the density of macromolecular structure during the stabilization process but also caused a certain degree of oxidative ablation. These led to a slight change on fibers’ surface of OPF. Through chemical pretreatment followed by stabilization, the obtained OPFHA-MEA had a relatively smooth surface in comparison with the OPF, as shown in Figure 5(c). This might be attributed to the catalysis effect of chemical reagents, which would prevent the fibers from generating a concentrated heat release during the following stabilization stage and thus avoid causing fibers’ defects and damages. For OPFHA-MEA-L fabricated in the order of chemical pretreatment, LbL assembly, and stabilization, it was found that the coating on the fibers’ surface was partially depleted, but it was still largely retained (Figure 5(d) to (g)). It meant that the CS-STPP flame-retardant coating still worked. The CS and STPP were assembled to form an intumescent flame-retardant system on the fibers’ surface.

SEM images of pristine PAN fibers and OPFs: (a) PAN, (b) OPF, (c) OPFHA-MEA, (d) OPFHA-MEA-5, (e) OPFHA-MEA-10, (f) OPFHA-MEA-15, and (g) OPFHA-MEA-20. SEM: scanning electron microscopy; PAN: polyacrylonitrile; OPF: polyacrylonitrile-based pre-oxidized fiber; MEA: monoethanolamine; HA: hydroxylamine hydrochloride.
XRD is used to characterize the crystalline structure, and the XRD patterns are illustrated in Figure 6. The diffraction peaks centered at 2θ angles of 17° and 29° for the pristine PAN fibers corresponded to (100) and (110) crystallographic planes of PAN fibers, respectively. 31 After stabilization in the air, there was no notable shift for the diffraction peak’s location at 17°, while the peak intensity significantly weakened, indicating that the PAN macromolecular chains were tended to be disordered. Meanwhile, a new diffraction peak appeared at 25°, which indicated that the original sequence structure was gradually destroyed and a new sequence structure was formed stepwise. This was because that PAN macromolecular chains were cyclized via dehydrogenation reactions and a six-membered heterocyclic structure with a large d-spacing was formed. Therefore, the area of the peak at 2θ = 25° could reflect the gradual accumulation of the cyclized structure formed by the PAN macromolecules during stabilization. It could be also seen from OPFHA-MEA and OPFHA-MEA-L curves, the diffraction peak at 2θ = 17° decreased and even disappeared while the diffraction peak at 25° widened. Combined with FTIR analyses (as shown in Figure 3), we knew that the conversion rate of –C≡N groups of PAN fibers was increased through chemical pretreatment. This would affect the contents of the stacked and/or cyclized fibers’ structures. At the same stabilization conditions, the degree of cyclization of the PAN fibers through chemical pretreatment with HA followed by MEA (i.e. PANHA-MEA and PANHA-MEA-L) was higher, which would inevitably affect the flame-retardant and mechanical properties of OPF.
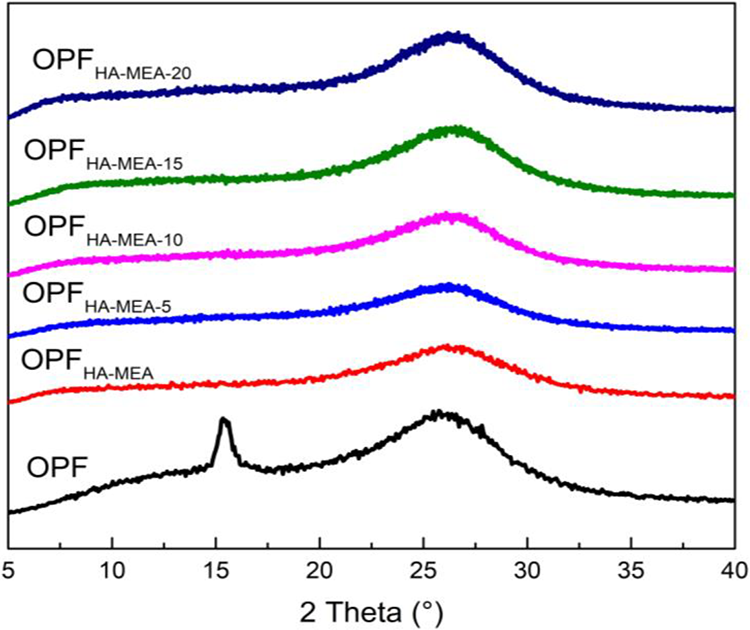
XRD patterns of OPFs (i.e. OPF, OPFHA-MEA, and OPFHA-MEA-L). XRD: X-ray diffractometer; OPF: polyacrylonitrile-based pre-oxidized fiber; MEA: monoethanolamine; HA: hydroxylamine hydrochloride.
Mechanical property
The mechanical property of pristine PAN fibers and OPFs is carried out, as shown in Figure 7, and the corresponding data are listed in Table 1. The breaking strength and elongation were about 375.6 N and 23.4%, respectively. However, after stabilization in air, the breaking strength and elongation of OPF, respectively, decreased to 220.3 N and 16.6%. The effects of different LbL assembly layers with CS and STPP on the mechanical property of OPF were also investigated. After chemical pretreatment with HA and MEA, the stabilization reactions’ temperature of the PAN fibers would decrease. In other words, the stabilization reactions occurred at a lower temperature for the PAN fibers. Therefore, the temperature of the oxidative exothermic reactions became wide and the concentrated exothermic phenomenon was alleviated. The damage of mechanical property of the fibers caused by the released concentrated exotherm was relieved, thus the obtained OPFs would have better mechanical property.
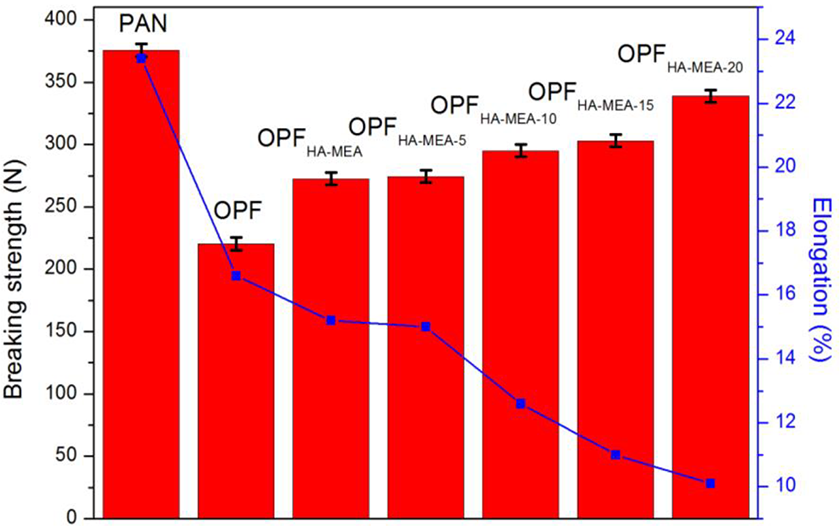
Mechanical property of pristine PAN fibers and OPFs (i.e. OPF, OPFHA-MEA, and OPFHA-MEA-L). PAN: polyacrylonitrile; OPF: polyacrylonitrile-based pre-oxidized fiber; MEA: monoethanolamine; HA: hydroxylamine hydrochloride.
Mechanical property and LOI values of pristine PAN fibers and OPFs.

LOI: limiting oxygen index; PAN: polyacrylonitrile; OPF: polyacrylonitrile-based pre-oxidized fiber; MEA: monoethanolamine; HA: hydroxylamine hydrochloride.
Through chemical pretreatment, LbL assembly, and stabilization in air, the breaking strength of the fibers was increased, as shown in Table 1. When CS-STPP flame-retardant coating reached up to 20 layers, the strength of OPFHA-MEA-20 increased, respectively, by 53.8% and 24.3% in comparison with OPF and OPFHA-MEA. The early stage of pre-oxidation was mainly the transformation of oxygen-containing structure. Meanwhile, the presence of flame-retardant coating played a certain insulation effect, avoided the direct contact between oxygen and fibers, and slowed the thermal damage of the pre-oxidation to the fibers. However, it did not affect the cyclization reaction, hence the fibers’ mechanical property was eventually increased.
Thermal stability
Thermal stability of the OPF, OPFHA-MEA, and OPFHA-MEA-L was investigated and evaluated by using TGA in a N2 atmosphere, as shown in Figure 8. It showed that all samples demonstrated the same degradation trend, and there were two steps in the degradation process. The fibers exhibited a weight loss at about 100°C, might be due to the degradation of the moisture absorbed and removal of chemical combined water. And the weight loss of OPFHA-MEA-L was slightly higher than that of OPF and OPFHA-MEA, might be attributed to the degradation of CS-STPP flame-retardant coating and its absorbed water. The second step happened when the temperature rose up to 400°C; such a weight loss was attributed to the dehydrogenation of PAN cyclized structure, which produced a graphite-like layer or ribbon structure. 32,33 It was also found from Figure 8 that the charred residue at 800°C for OPF was about 59.9 wt%, and was slightly lower than that of OPFHA-MEA (about 61.4 wt%). But for OPFHA-MEA-L (i.e. OPFHA-MEA-20), the presence of the intumescent CS-STPP flame-retardant coating greatly increased the charred residue and it reached up to 72.1 wt%. This might be that after chemical pretreatment, the exothermic reactions of the fibers were more moderate, the stability of the carbon–nitrogen conjugated ring structures was promoted and the thermal degradations of the fibers were relatively reduced. Meanwhile, STPP released polyphosphoric acid and PA compounds, which might react with CS to form a crosslinked carbon layer during thermal degradation. All of the factors inhibited the decomposition of the fibers and increased the amount of charred residue, contributing to the improvement of thermal stability.
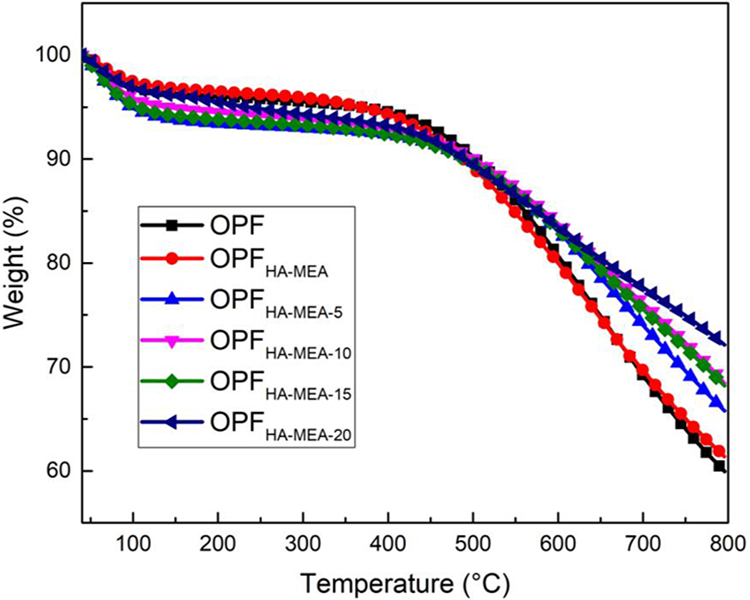
TGA curves of OPF, OPFHA-MEA, and OPFHA-MEA-L. TGA: thermogravimetric analysis; OPF: polyacrylonitrile-based pre-oxidized fiber; MEA: monoethanolamine; HA: hydroxylamine hydrochloride.
Flame-retardant property
The LOI of the pristine PAN fibers and OPFs is tested and the results are also listed in Table 1. After pre-oxidation, the PAN fibers underwent cyclization, dehydrogenation, and oxidation reactions. These reactions would convert the original linear macromolecular structures into heat-resistant planar trapezoidal structures, and thus increased the LOI values of OPF. The LOI value of PAN fiber was only 18.0%, and belonged to highly flammable fibers. After stabilization in air, the LOI value of OPF rose up to 25.7%. The LOI value of the OPFHA-MEA reached 37.3%, increased by 31.1% compared to that of OPF. This was because chemical pretreatment caused chemical reactions of functional groups (e.g. cyano and ester groups) on the macromolecular chains to produce new groups. The presence of new groups improved the thermal property of PAN fibers, decreased the initial pre-oxidation reaction temperature, and accelerated the pre-oxidation reaction rate. These aspects contributed to higher LOI value of fibers.
Meanwhile, Table 1 also showed that the LOI value of the OPFHA-MEA-L increased with the increase of CS-STPP LbL assembly layers. In comparison with OPFHA-MEA (without CS-STPP flame-retardant coating), the LOI values of the corresponding OPFHA-MEA-L (L = 5, 10, 15, and 20) were increased to 38.8%, 41.5%, 42.4%, and 44.6%, respectively. The improvement of flame-retardant property was attributed to the high efficiency of the CS-STPP layer in hindering the transportation of heat and oxygen. The CS and STPP were employed to fabricate intumescent flame-retardant coating on the fibers’ surface, and the synergistic inhibitory effects of N and P elements protected fibers from burning off. During combustion, CS could be carbonized to hinder the combustion, and at the same time, nontoxic, noncorrosive, and nonflammable gases such as CO2, N2, and NH3 were released. STPP was thermally decomposed into polyphosphoric acid and PA compounds with strong dehydration function during heating, which took part in the dehydration of the CS and yielded the carbonaceous and phosphocarbonaceous residue. The released gaseous substances made the mixtures of the carbonaceous and phosphocarbonaceous residue to swell, and led to the formations of the intumescent charred residue.
Conclusion
In this work, novel high-performance OPFs were fabricated through combining the techniques of chemical pretreatment with HA and MEA, LbL assembly with environmentally friendly intumescent CS-STPP flame-retardant coating, and stabilization in air. The morphological structure, thermal stability, and flame-retardant and mechanical properties of the fabricated OPFs were studied. The results indicated that PAN fibers were successfully modified by chemical pretreatment with HA and MEA, which provided the condition for following LbL assembly. The crystalline structure of the fibers happened to change due to the occurrences of cyclization reactions during the stabilization process. The fabricated OPFs demonstrated excellent thermal stability and flame-retardant and mechanical properties, that is, the obtained OPFHA-MEA-10 fibers had superior comprehensive properties with charred residue of about 68.2%, breaking strength of about 295.1 N, breaking elongation of 12.6%, and LOI value of about 41.5%, respectively. It was envisioned that the fabricated OPFs had potential applications such as high temperature filtration, composites, high-performance fibers and its textiles.
Footnotes
Author contributions
Xiaolu Sun and Songqi Li contributed equally to this work.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Equipment Pre-research Joint Fund of Ministry of Education (6141A02022267), Fundamental Research Funds for the Central Universities (JUSRP51621A, JUSRP52007A, and JUSRP41910), the Open Project Program of Jiangsu Advanced Textile Engineering Technology Center (XJFZ/2018/04), and the Undergraduate Innovation and Training Program of Jiangnan University (2020212Y).