
Abstract
Bio-based phthalonitrile resins were synthesized using a two-step process. Initially, vanillin underwent a nucleophilic reaction with nitro and potassium carbonate to form intermediates, which were then dissolved in N, N-dimethylformamide. The intermediate was divided into two portions, with 4,4’-oxydianiline and 4,4’-methylenedianiline added to each portion, respectively, to produce two different vanillin-based phthalonitrile monomers: OVA monomer and DVA monomer. The successful synthesis of these two bio-based phthalonitrile monomers was confirmed using Fourier transform infrared spectroscopy and proton nuclear magnetic resonance spectroscopy. Differential scanning calorimetry was employed to study the processability of the monomers, revealing that OVA monomer and DVA monomer had a wide processing window of 110°C and 69°C, respectively, which were advantageous for resin processing. Dynamic mechanical analysis showed that the storage modulus of the OVA and DVA resins reached 4818 MPa and 3526 MPa, respectively, with a 24-h water absorption rate of only 1.03% and 0.90%. This study presents a viable method for synthesizing bio-based phthalonitrile resins, which can be widely utilized as functional and structural materials in daily life due to their renewability, degradability, and minimal environmental impact.
Introduction
Among previous developments, phthalonitrile (PN) resins stand out as particularly attractive, widely studied, and extensively applied in industries such as aerospace, aircraft, automotive, microelectronics, and marine sectors.1,2 Originally developed by the United States Naval Research Laboratory, phthalonitrile is a high-temperature resin expected to serve as a high-performance material capable of replacing metal components as a polymer matrix material. Phthalonitrile resins offer several advantages, including high thermal stability, excellent resistance to thermal oxidation, superior moisture resistance, good flame retardancy, and strong mechanical properties. The presence of curing agents with aromatic structures within the resin contributes to a high degree of cross-linking and thermal stability. Additionally, the abundance of aromatic structures allows phthalonitrile resins to be used as matrices for radiation shielding, UV protection, and for glass and carbon fiber-reinforced composites, which can operate at temperatures as high as 400°C.3–6 However, most phthalonitrile monomers are synthesized through nucleophilic substitution reactions between phenol and 4-nitrophthalonitrile.7,8 These monomers are typically derived from petroleum-based substances, such as bisphenol A, bisphenol F, and resorcinol, which are not only non-recyclable but also contribute to significant environmental pollution.9–11 Moreover, phthalonitrile polymers synthesized from petroleum-based feedstocks tend to have high melting points and narrow processing windows, significantly limiting their processing conditions and hindering their expansion into other industrial applications. In response to these challenges, the synthesis of phthalonitrile from biomass-based feedstocks has emerged as a promising alternative. 12 Biomass materials are renewable, abundant, and cost-effective, offering the added benefits of being degradable and environmentally friendly.
Resveratrol (trans-3,5,4’-trihydroxystilbene), a natural product derived from biomass, is a renewable feedstock and a less toxic alternative to bisphenol in phthalonitrile (PN) systems. Additionally, resveratrol’s unique structure can enhance the thermosetting properties of PN resins. Incorporating resveratrol into PN resins has the potential to address the toxicological issues associated with current resin technologies. This approach involves synthesizing a new type of PN resin through the nucleophilic reaction of renewable triphenols. The resulting resin exhibits excellent viscosity parameters comparable to those of current high-temperature resins, while also widening the processing window, thereby improving processing performance. The new PN resin features no glass transition temperature (Tg), excellent thermal oxidation resistance, a low dielectric constant, and low water absorption after curing. 13
Typically, the melting temperatures (T ms ) of petroleum-based phthalonitrile monomers are around 200°C.10,14 However, in the synthesis of bisfunctional phthalonitrile monomers containing allyl/propargyl ether groups from sustainable vanillin, the presence of flexible allyl/propargyl ether and methoxy groups reduces molecular rigidity and intermolecular interactions, thereby lowering the T ms . As a result, these monomers exhibit a lower T ms and a wider processing window, with the cured polymers demonstrating excellent thermal stability in a nitrogen atmosphere. 15
A bio-based phthalonitrile resin precursor was successfully synthesized by gradually derivatizing four phenolic hydroxyl groups with 4-nitrophthalonitrile (NPh) using catechin (CA) as the raw material. Compounds containing unreacted phenolic groups enable a “self-curing” polymerization process at high temperatures, resulting in a resin with high heat resistance. This CA-Ph system does not require the addition of any external curing agents and offers better thermal stability compared to petroleum-based BPA-Ph resin formed by reacting bisphenol A with NPh, which typically includes 5% 4,4’-diaminodiphenyl sulfone (DDS) as an initiator. Additionally, this system exhibits a lower melting point and curing temperature, further improving processing performance.13,16
Furthermore, bio-based phthalonitrile precursors were synthesized via a one-step process using natural honokiol derivatives as raw materials. These precursors can undergo a self-curing reaction with the allyl group without sacrificing the thermal stability of the thermoset resin. Among the two bio-based precursors, sandalwood phenolic derivatives demonstrate a wide processing window of 163°C and a melting point below 70°C. After curing, the glass transition temperature of honokiol and honokiol-based resin exceeds 500°C, and the 5% weight loss temperature reaches 527°C. 17 These properties surpass those of the allyl-free petroleum-based comparator, further highlighting the potential of new thermosetting resins prepared from biomass-based materials as promising alternatives.
Significant progress has been made in the preparation of thermosetting resins, particularly epoxy resins and cyanate materials, using bio-based materials. These bio-based resins exhibit properties comparable to those of petroleum-based compounds and offer broad application prospects.18–20 However, in the realm of phthalonitrile resins, the variety of available phenols is limited. Among the available phenolic compounds, vanillin, a renewable biosynthetic molecule, shows promise as a molecule that can be synthesized with phthalonitrile. The resulting novel phthalonitrile resin exhibits excellent fluid viscosity and, upon complete curing, demonstrates excellent thermal stability while maintaining its intact structure. These results indicate that the phthalonitrile resin synthesized from vanillin possesses the dual advantages of high-temperature and mechanical stability. 21 In addition, eugenol from biomass reacts with 4-nitrophthalonitrile in an environmentally friendly solvent through a nucleophilic substitution reaction, and no small molecular catalyst is needed during the curing process to produce bio-based acrylate-derived phthalonitrile monomers. This study indicates that both resins have good processing properties, and both exhibit high glass transition temperatures. 22 These researches significantly enhances the effective utilization of biomass, molecular design, and environmentally friendly solutions for bio-based synthetic polymer materials. 23
Based on the aforementioned research, this study further research on the performance of vanillin-type phthalonitrile resin was conducted using different curing agents. Biomass-derived vanillin was used in a nucleophilic substitution reaction with 4-nitrophthalonitrile (4-NO2) to prepare two biomass vanillin-type phthalonitrile monomers. These monomers, 4-(4-((Z)-((3-(4-(((Z)-4-(4,5-dicyano-2-methoxyphenoxy)benzylidene)amino)phenoxy) phenyl) imino)methyl)phenoxy)-5-methoxyphthalonitrile (OVA) and 4-(4-((Z)-((3-(4-(((Z)-4-(3,4-dicyanophenoxy)-3-methoxybenzylidene) amino) benzyl) phenyl) imino) methyl)phenoxy)-5-methoxyphthalonitrile (DVA), were synthesized and characterized through a two-step method. The curing behavior, thermal stability, processing properties, and mechanical properties of these bio-based phthalonitrile resins were thoroughly investigated. The results demonstrated that bio-based materials can achieve properties comparable to petroleum-based phthalonitrile resins, making them suitable for long-term use and even functional materials in everyday life. This study provides a feasible design strategy for the diversified synthesis of biomass-based phthalonitrile resins, expanding the potential applications and sustainability of these materials.
Experiments
Materials
N, N-dimethylformamide (DMF, 99.5%) was bought from Tianjin Jingdong Tianzheng Fine Chemical Reagent Factory, was analytically pure and used without further purification. 4-Nitrophthalonitrile (98%) was supplied by Shanghai Macleane Biochemical Technology Co., Ltd and used without further purification. Anhydrous potassium carbonate (K2CO3, 99.0%) was purchased from Tianjin Fengchuan Chemical Co and used as received. 4,4’-oxydianiline (ODA, 98%) was purchased from Shanghai Macleane Biochemical Technology Co., Ltd, and was analytically pure. We used it without further purification. 4,4’-methylenedianiline (DDM) was bought from Beijing Chemical Plant, and was analytically pure and used without further purification. Absolute ethanol (C2H6O), analytical pure, was supplied by Tianjin Yongda Chemical Reagent Co., Ltd.
Synthesis
The bio-based phthalonitrile monomers were prepared by nucleophilic substitution, as shown in Figure 1. Firstly, 0.031 mol of vanillin and 0.032 mol of anhydrous potassium carbonate, 40 mL of DMF were to added the three-mouth flask, heated the oil bath at 80∼85°C and stirred to dissolve the reaction for 1 h. Then gradually add 0.023 mol of 4-nitrophthalonitrile was gradually added and stirred for 8 h. After the reaction was over, a certain amount of deionized water was poured into the product at 25∼30°C, and repeated three times. The obtained product was cleaned with distilled water and then put into a mixed solution of ethanol and water 4:1 for washing, repeated three times, and then obtained the bio-based phthalonitrile intermediate, 4-(4-formyl-2-methoxyphenoxy)phthalonitrile (FVA). Secondly, 0.005 mol of 4,4’-oxydianiline (ODA) and 0.005 mol of 4,4’-methylenedianiline (DDM) were added to the three-mouth flask containing 0.011 mol of FVA and 20 mL of DMF, respectively, and stirred in an oil bath at 80∼85°C. After the reactions were over, the products were poured into deionized water until particles were precipitated. The crude products were obtained by filtration, and then stirred and heated at 80°C in a 4:1 mixed solution of ethanol and water, repeated three times, and then obtained the bio-based phthalonitrile monomers: OVA and DVA. Synthetic route of the FVA(a), OVA(b1), DVA(b2).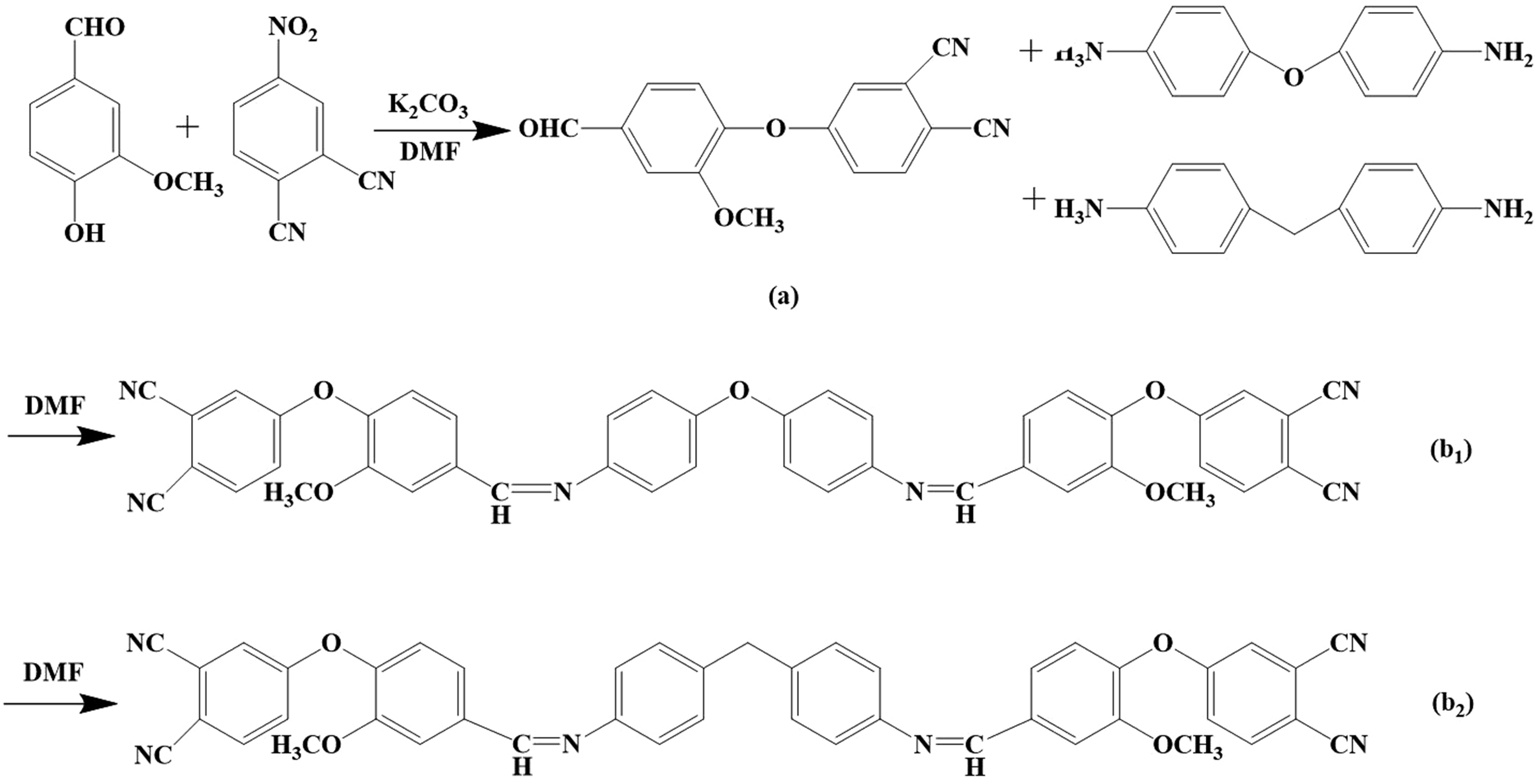
Curing process of samples
Taking the curing process of OVA as an example, the curing process of OVA was the same as described below. The 2.000 g of OVA monomer was put into a 45 mm × 12 mm open tin foil mold, the tin foil was glued to the iron sheet with high-vacuum silicone grease, and then the iron sheet was placed in a vacuum drying oven and heated to 210°C. After the monomer was melted, the 0.040 g of curing agent ODA was added and degassed, and the OVA monomer bubbles can be observed to change from large to small, and the system gradually became viscous and then stopped degassing. The sample was then placed in the muffle furnace for post-curing treatment, which was divided into 4 stages: 200°C for 3 h, 230°C for 3 h, 260°C for 3 h, 290°C for 3 h, and 320°C for 3 h. The OVA resin was machined into rectangular specimens (40 mm × 10 mm × 2 mm) for dynamic mechanical analysis (DMA) experiments, and the powder of the OVA resin was used for thermogravimetric analyses (TGA).
Characterization
The Fourier Transform Infrared (FT-IR, AVATAR380, Nigolis Inc., USA) spectra of the FVA monomer, the OVA monomer, and the DVA monomer were recorded using KBr pellets between 4000 and 400 cm−1 in air. The structural changes within the monolith of the FVA monomer, the OVA monomer, and the DVA monomer were observed using nuclear magnetic resonance hydrogen spectrometer (1H NMR, Bruker 400 MHz Advance; 13C NMR, Bruker 101 MHz Advance, Bruker Biotechnology Co., Ltd. in Germany, DMSO-d6). The elemental content of the monomers was observed by organic element analyzer (EA, Elementar Unicube, German). The melting points and the curing temperatures of the OVA monomer and the DVA monomer were tested using differential scanning calorimetry (DSC, DSC 214 Polyma, NETZSCH Instrument Manufacturing GmbH, Germany). The test temperature is 50∼370°C, the heating rate is 10°C/min, the gas flow rate is 15 mL/min, and the nitrogen atmosphere is used. The thermal stability of the OVA resin and the DVA resin was tested by the thermogravimetric analyzer (TGA, STA 449C NETZSCH Instrument Manufacturing GmbH in Germany). The test temperature is 50∼800°C, the heating rate is 10°C/min, the gas flow rate is 15 mL/min, and the nitrogen atmosphere is used. To test the water absorption of the OVA resin and the DVA resin, the samples were soaked in deionized water at 25°C, and the samples were taken out dried and weighed every 2 h, and soaked for a total of 24 h to observe the weight change. To study the dynamic mechanical properties of the OVA resin and the DVA resin, the storage modulus and glass transition temperature (Tg) were tested using a dynamic thermomechanical analyzer (DMA, Q800, United States TA). The length, width, and thickness of the sample are 30 mm × 10 mm × 2 mm, the test temperature is 50∼400°C, the heating rate is 5°C/min, and the test frequency is 1 Hz.
Results and discussion
Design and synthesis of phthalonitrile monomers
Figure 2 presents the FT-IR spectra of FVA, OVA, and DVA. For FVA, the C-H stretching vibration of the -CHO group was observed at 2842 cm−1, the -C = O vibration of the -CHO group appears at 1702 cm−1, and the -CN group was identified by the peak at 2231 cm−1. The hydroxyl group in vanillin reacted with the nitrile group in 4-nitrophthalonitrile to form an ether bond, evidenced by the aromatic ether bond stretching vibration at 1245 cm−1, which is present in the bio-based phthalonitrile intermediate (FVA). For OVA, the FT-IR spectrum showed a stretching vibration peak for unsaturated C-H at 3020 cm−1, while the stretching vibrations of the benzene ring skeleton were observed at 1600 cm−1, and 1480 cm−1. The -CN absorption peak was present at 2230 cm−1, and the aromatic ether bond stretching vibration is noted at 1240 cm−1. The monomer synthesis involves the dehydration of aldehyde and amine groups to form imine bonds, which was confirmed by the disappearance of the aldehyde peak at 1700 cm−1 and the appearance of the imine bond peak at 1630 cm−1 in the spectrum. For DVA, the stretching vibration peak for unsaturated C-H was found at 3035 cm−1, with the benzene ring skeleton showing stretching vibrations at 1587 cm−1 and 1486 cm−1. The -CN absorption peak appeared at 2233 cm−1, and the aromatic ether bond stretching vibration was observed at 1243 cm−1. FT-IR spectra for FVA(a), OVA (b1), and DVA (b2).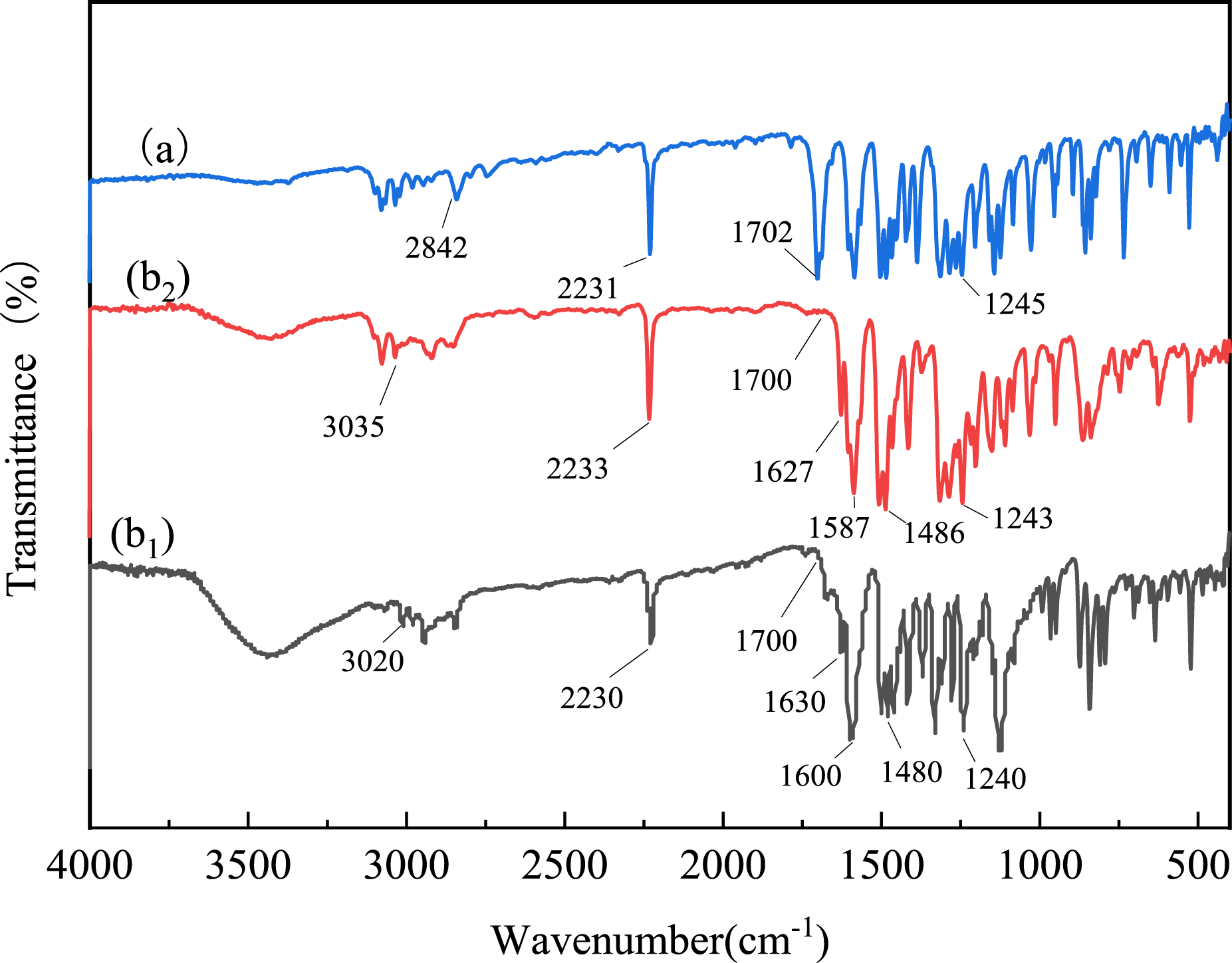
Figure 3 shows the 1H NMR spectrum of FVA monomer. The specific chemical shifts are as follows: 1H NMR (400 MHz, DMSO-d6, δ):10.02 (s, 1H), 8.09 (d, J = 8.8 Hz, 1H), 7.78 (d, J = 2.7 Hz, 1H), 7.71 (d, J = 1.9 Hz, 1H), 7.66 (dd, J = 8.0, 1.9 Hz, 1H), 7.45 (d, J = 8.1 Hz, 1H), 7.37 (dd, J = 8.7, 2.7 Hz, 1H). 1H NMR spectra of FVA monomer.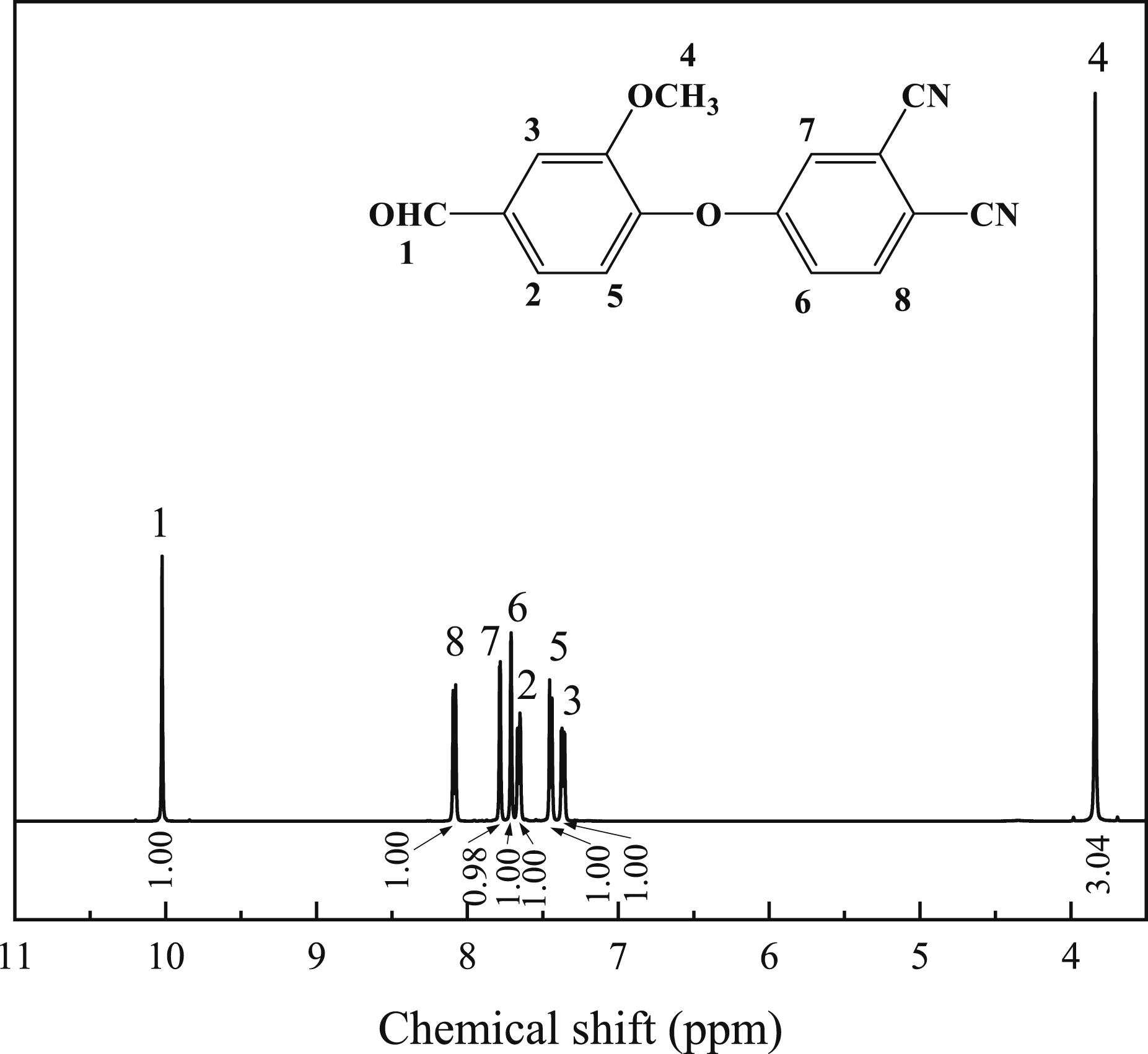
The 1H NMR and 13C NMR spectra of OVA monomer is shown in Figure 4. The NMR hydrogen spectroscopy was analyzed for specific chemical shifts as follows:1H NMR (400MHZ, DMSO-d6, δ): 8.71 (s, 1H), 8.09 (d, J = 8.8 Hz, 1H), 7.82 (d, J = 1.8 Hz, 1H), 7.76 (d, J = 2.6 Hz, 1H), 7.62 (dd, J = 8.3, 1.7 Hz, 1H), 7.39 (dd, J = 8.5, 2.4 Hz, 2H), 7.35 (d, J = 2.6 Hz, 1H), 7.33 (d, J = 2.7 Hz, 1H), 7.13 (d, J = 8.4 Hz, 2H), 3.85 (s, 3H). 13C NMR (101 MHz, DMSO-d6, δ): 160.86, 158.95, 155.21, 151.26, 146.60, 143.55, 136.11, 135.24, 122.96, 122.75, 122.58, 121.33, 120.71, 119.29, 116.54, 115.87, 115.35, 112.12, 107.93, 55.88. Elemental Analysis: C 72.95, H 3.92, N 11.66, O 11.47. 1H NMR and 13C NMR spectra of OVA monomer.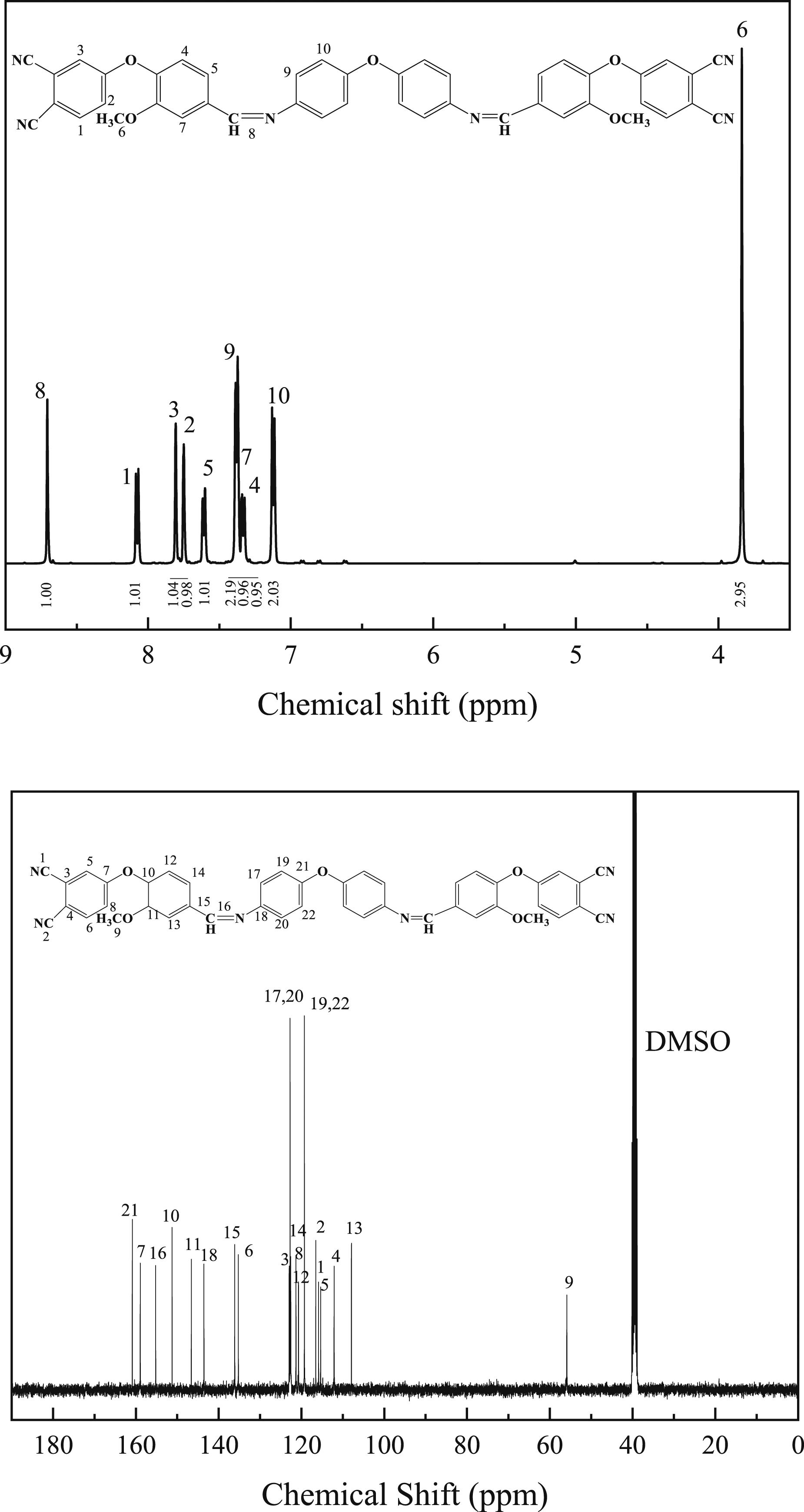
The 1H NMR spectra of DVA monomer is shown in Figure 5. The NMR hydrogen spectroscopy was analyzed for specific chemical shifts as follows:1H NMR (400MHZ, DMSO-d6, δ): 8.68 (s, 1H), 8.09 (d, J = 8.8 Hz, 1H), 7.80 (d, J = 1.8 Hz, 1H), 7.77 (d, J = 2.6 Hz, 1H), 7.60 (dd, J = 8.2, 1.8 Hz, 1H), 7.38 (d, J = 8.2 Hz, 1H), 7.35 (d, J = 2.3 Hz, 1H), 7.33 (d, J = 1.9 Hz, 2H), 7.26 (d, J = 8.2 Hz, 2H), 4.03 (s, 1H), 3.84 (s, 3H). 13C NMR (101 MHz, DMSO-d6, δ): 160.84, 159.13, 151.23, 149.14, 143.52, 139.36, 136.10, 135.21, 129.48, 122.96, 122.55, 121.32, 121.12, 120.69, 116.51, 115.85, 115.33, 112.07, 107.88, 55.86. In the carbon spectrum, the chemical shift values of the carbons in the methylene group overlap with those of the carbons in the solvent DMSO, and therefore cannot be observed. Elemental Analysis: C 74.93, H 4.24, N 11.72, O 9.11. In summary, the results of infrared, nuclear magnetic resonance and organic element analyzer can be used to determine the successful preparation of the target product. 1H NMR and 13C NMR spectra of DVA monomer.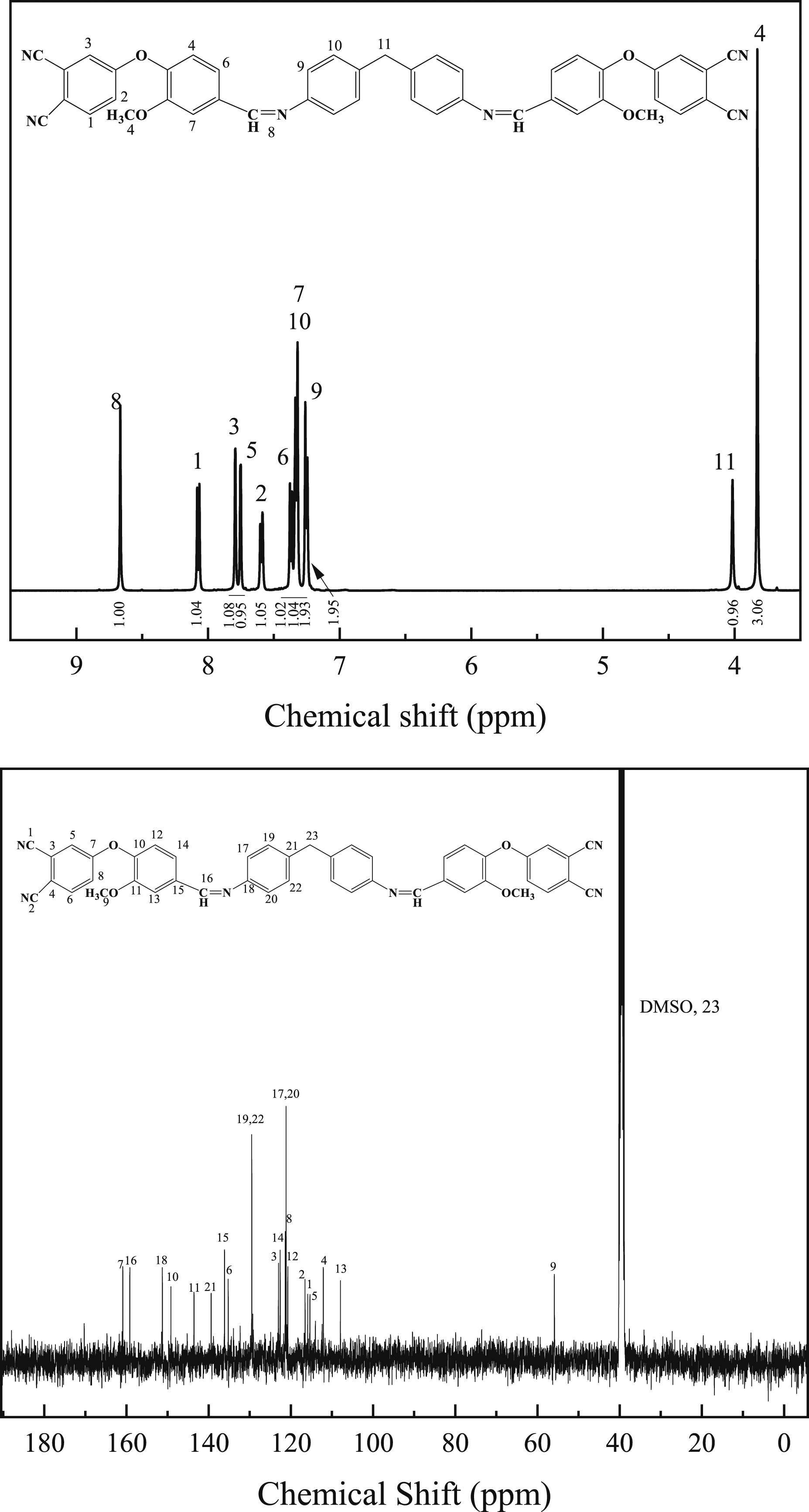
Curing behavior and processability
DSC curves for the OVA monomer and the DVA monomer are presented in Figure 6. Among the two monomers, OVA exhibited a lower melting point of 179°C, likely due to the presence of flexible ether bonds within its molecular structure. In contrast, DVA melts at a higher temperature, above 230°C, which poses challenges for the vacuum degassing process in a vacuum drying oven and may hinder the preparation of dense plates. Additionally, OVA had a processing window of 110°C, while DVA’s processing window was narrower at 69°C. The wider processing window of OVA makes it more suitable for plate preparation. Both monomers demonstrate high decomposition temperatures, with OVA decomposing at 354°C and DVA at 351°C. These high decomposition temperatures indicate that both OVA and DVA possess good processing performance at elevated temperatures. DSC curves for the OVA monomer and the DVA monomer, (a) no curing agent, (b) added curing agent.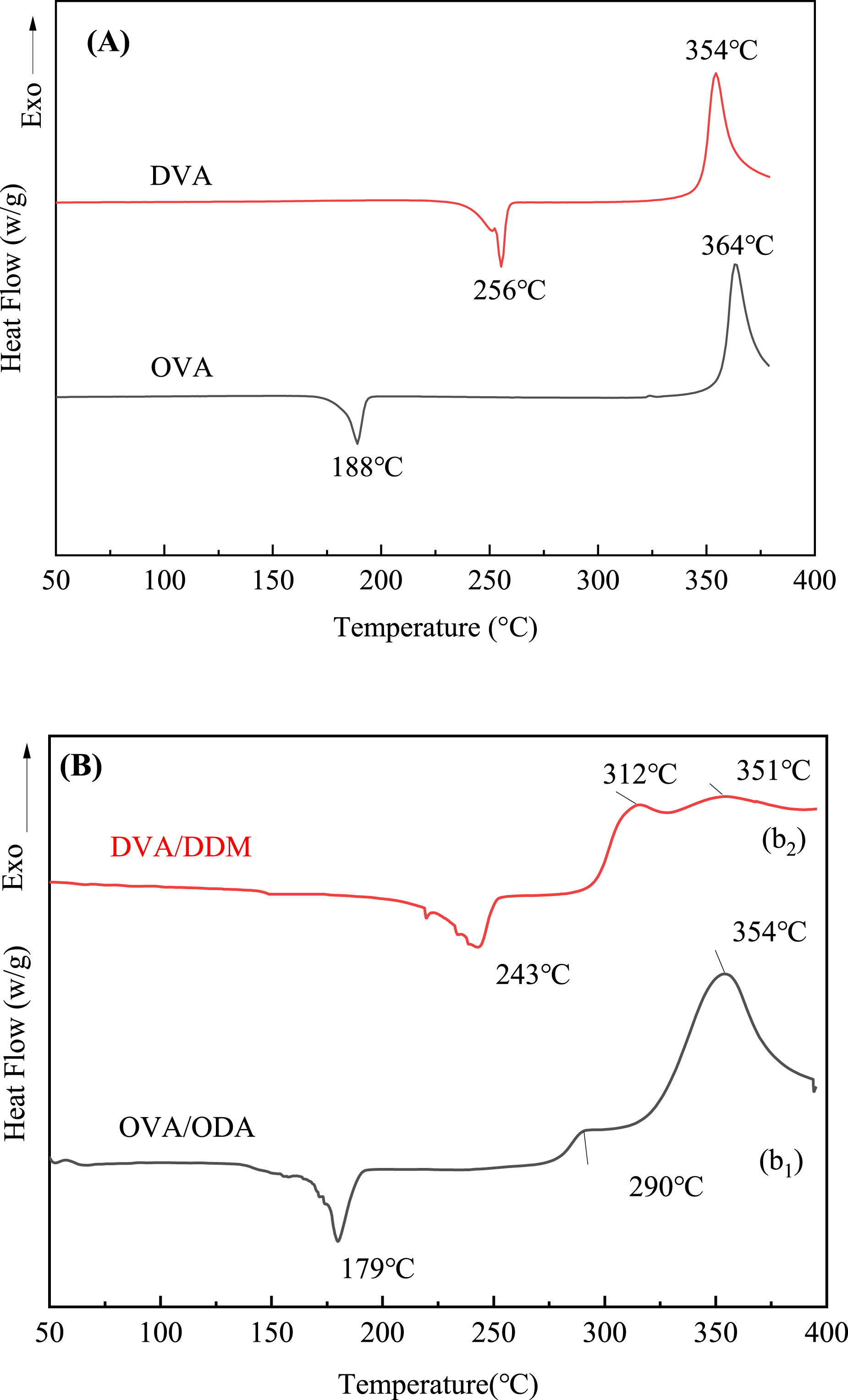
Thermal stability
The thermal analyzer was used to assess the thermal stability of the OVA and DVA resins, with the thermogravimetric curves shown in Figure 7. The data indicate that at the same thermal weight loss, both OVA and DVA resins retained 94.7% of their weight. At 350°C, the thermal weight of the OVA resin was 93.3%, while the DVA resin retained 92.9% of its weight. This suggests that both OVA and DVA resins experience less than 5% thermal weight loss when approaching 300°C. Specifically, at 350°C, the OVA resin exhibited a thermal weight loss of 6.7%, compared to 7.1% for the DVA resin. Furthermore, the temperature at which 10% of the thermal weight loss occurred was above 400°C, indicating a high initial decomposition temperature. Thermal stability of the OVA resin is found to be superior to that of the DVA resin, likely due to the presence of an additional ether bond in the OVA resin. Ether bonds are more resistant to degradation, contributing to the better thermal stability observed in the OVA resin. TGA curves of the OVA resin (A) and DVA resin (A).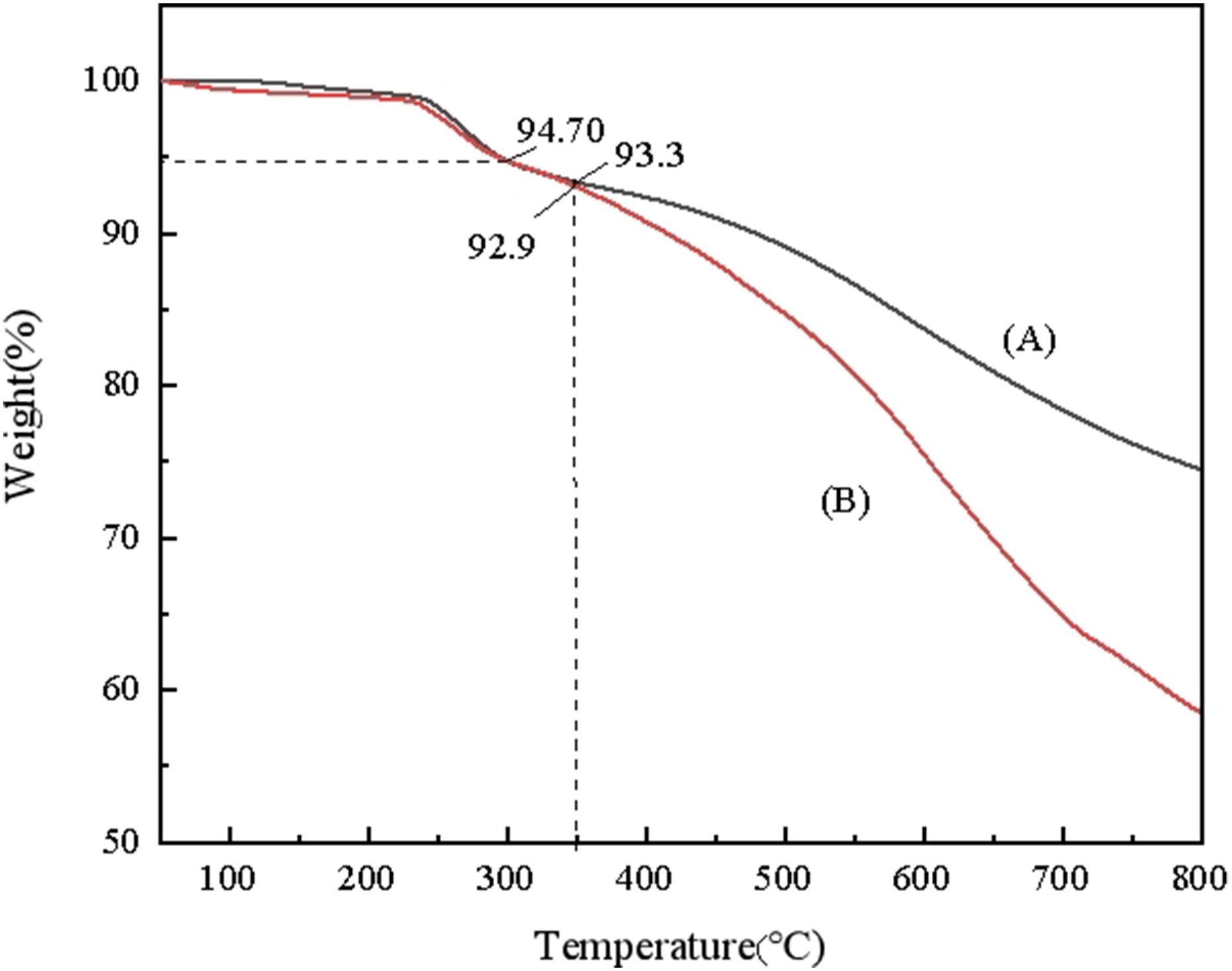
Dynamic mechanical analysis
The resins were subjected to DMA testing to investigate the effect of the structure of phthalonitrile resins on the storage modulus and glass transition temperature, and the results are shown in Figure 8. The storage modulus reflected the rigidity of the material, and the Tg could be determined from the temperature corresponding to the peak of the Tan Delta and the temperature curve. As shown in Figure 8(A), the OVA resin exhibited an excellent storage modulus of up to 4818 MPa. Additionally, a stray peak appears in the Tan Delta curve at 364°C, which may be attributed to decomposition, as supported by infrared analysis. Figure 8(B) shows that the DVA resin also has a good storage modulus, reaching 3526 MPa. A stray peak in the Tan Delta curve at 351°C may also indicate decomposition, and like the OVA resin, no clear glass transition temperature was observed. The lower storage modulus of the DVA resin compared to the OVA resin could be due to the structural differences between the two resins. The melting point and cross-linking point of OVA monomers containing flexible ether bonds are lower. Under the same catalyst, OVA monomers are more likely to undergo cross-linking reactions than DVA monomers, resulting in a higher degree of cross-linking of OVA resins than DVA resins. DMA plots for the OVA resin (A) and the DVA resin (B).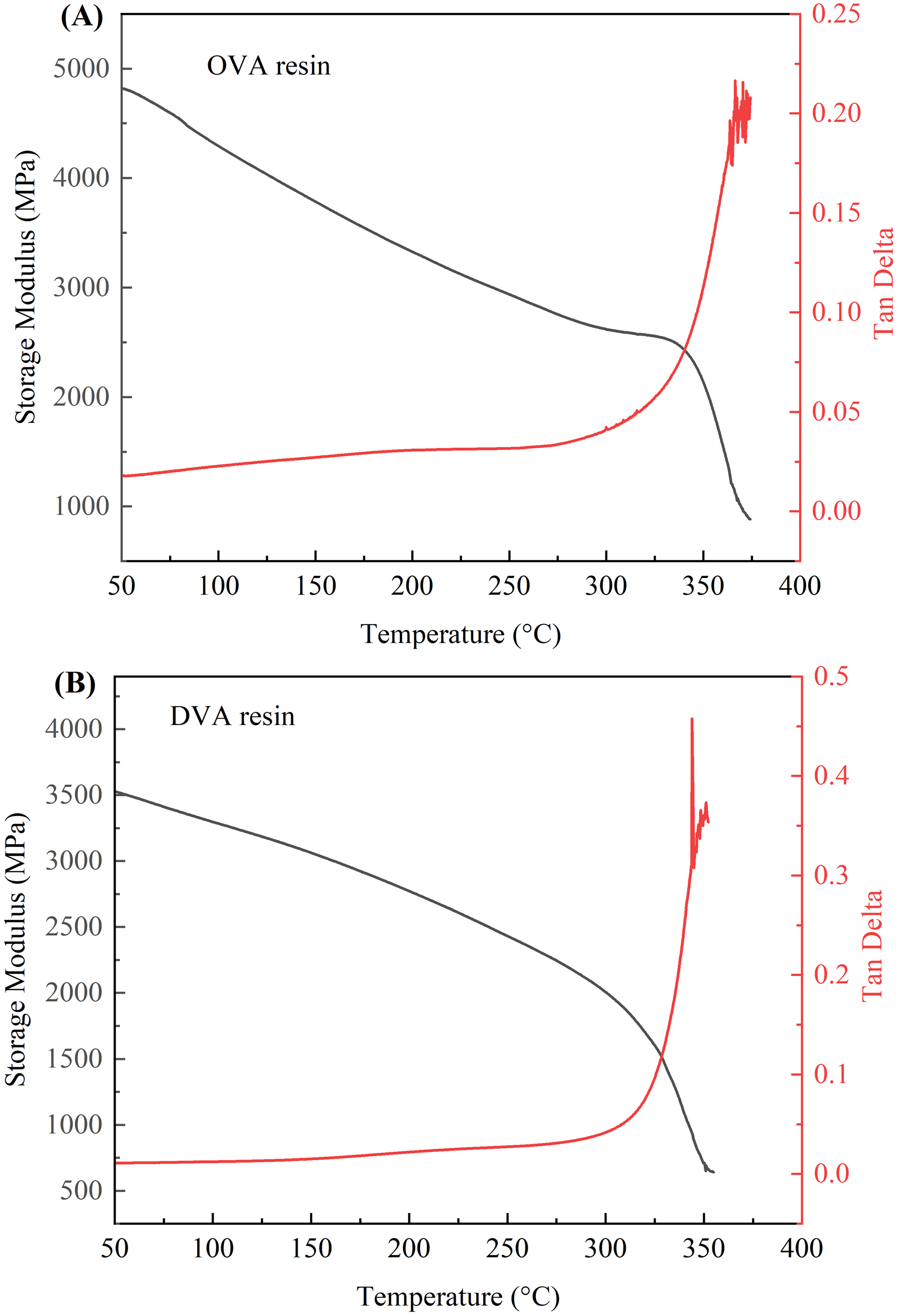
Water absorption properties
The water absorption of the OVA resin and the DVA resin was studied, and the results are shown in Figure 9. The water absorption rate of the resins increased rapidly in the first 14 h, stabilized after 20 h, and stayed at 1.03% and 0.90% after 24 h, which indicates that the resins have a certain hydrophilicity. The water absorption rate of OVA resin is slightly higher than that of DVA resin, which can be attributed to the presence of an ether bond in the OVA resin. The oxygen atom in the ether bond has a high electronegativity, leading to an increase in hydrophilicity. However, with the increase of time, the water absorption noticeably decreases, possibly due to the high crosslinking density that hinders the penetration of water molecules into the polymer matrix. Water absorption rates of the OVA resin (A) and the DVA resin (B).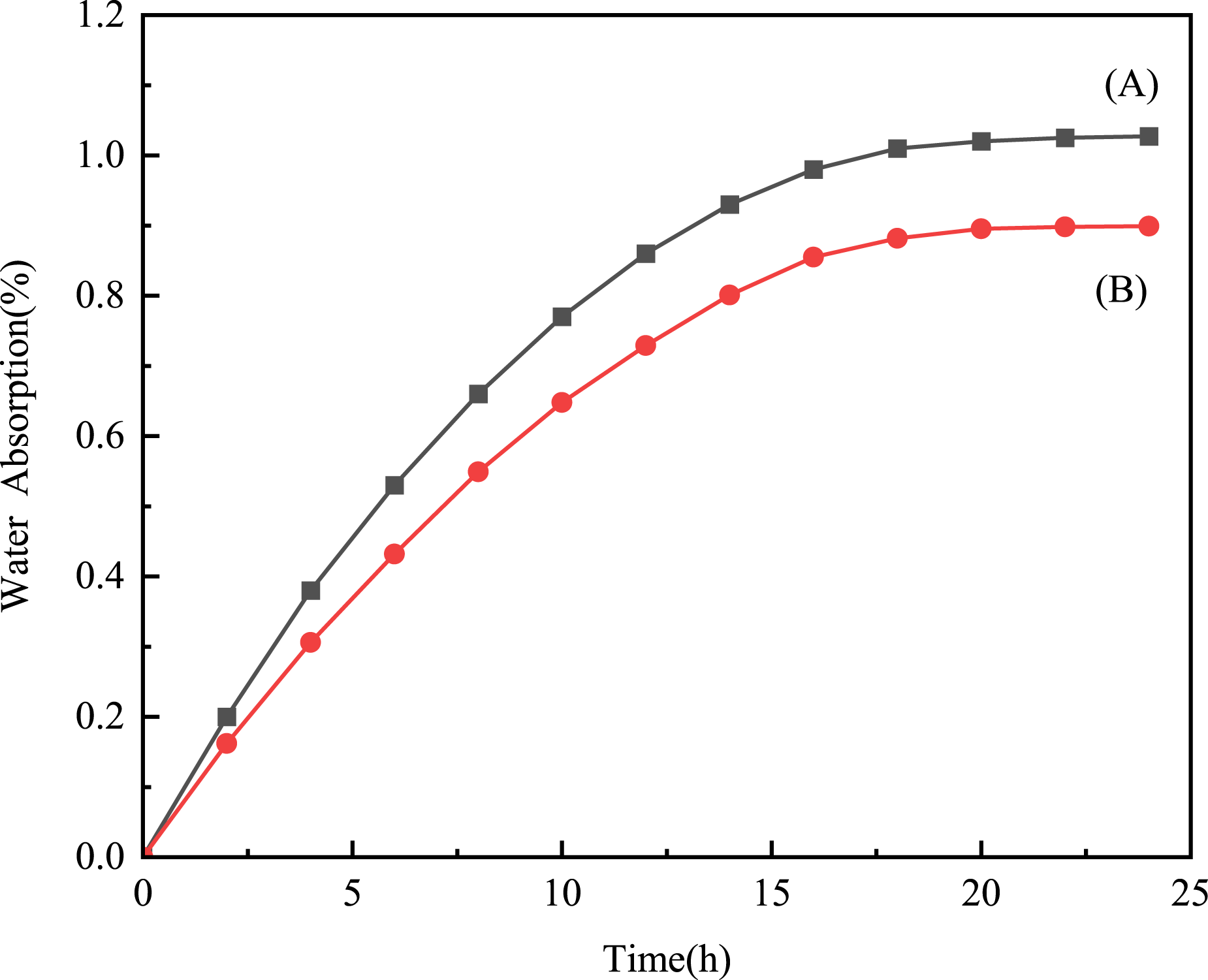
The formula for calculating water absorption (A) is as follows.

Conclusion
In summary, OVA and DVA monomers were synthesized using bio-based vanillin as the raw material through nucleophilic reactions, with the route designed to reduce costs and minimize pollution. The chemical structure of the resulting monomer was also confirmed by 1H NMR, and FT-IR spectroscopy. Their machinability and thermal stability were studied and discussed using DSC and TG analysis. The results showed that OVA exhibited a wider processing window compared to DVA. Additionally, the introduction of the vanillin structure improved processing performance without compromising thermal stability. This work provided a novel approach to producing thermoset materials from biomass feedstocks and supports the feasibility of developing other bio-based thermoset materials. Dynamic mechanical analysis revealed that the OVA resin achieved a maximum storage modulus of 4818 MPa, while the DVA resin reached 3526 MPa, demonstrating excellent mechanical properties. These findings suggest that bio-based phthalonitrile polymers have significant potential for use in aerospace, microelectronics, and other applications requiring high-performance bio-based materials.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was financially supported by the Natural Science Foundation of Hebei Province, China (B2023209044).
Data availability statement
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.