
Abstract
Phthalonitrile (PN) resins exhibit outstanding thermal and mechanical properties but suffer from intrinsically high melting temperatures and sluggish curing kinetics, which severely restrict their processability and manufacturability. In this work, a eutectic blending strategy based on ternary PN monomers was employed to markedly depress the melting temperature, enabling mild and controllable melt prepolymerization with a diamine curing agent. This approach produced a low-melting phthalonitrile prepolymer (MPPh) exhibiting a viscosity below 1 Pa·s at 88°C, providing a stable melt-processing window. DSC and FTIR analyses revealed that the prepolymerization predominantly proceeded through linear chain growth via amino-nitrile addition reactions. To address the inherently low curing reactivity of MPPh, 3-aminophenylacetylene (APA) was introduced as a reactive diluent to activate the curing process and further tailor the rheological behavior. The modified resin (MP-m) demonstrated significantly enhanced curing efficiency and a reduced viscosity below 1 Pa·s at 66°C, excellent isothermal stability, and pronounced shear-thinning behavior. The cured MP-m resin retained excellent thermal stability and mechanical performance. These findings provide a simple and solvent-free strategy for the preparation of low-melting-point phthalonitrile, broadening the practical application potential of PN resins in high-temperature composite manufacturing.
Introduction
Phthalonitrile resins (PN) represent an important class of high-performance thermosetting polymers and have attracted growing research interest in recent years.1–5 Their curing process involves the transformation of nitrile groups into phthalocyanine, triazine, and isoindoline structures, leading to highly cross-linked, void-free networks composed of thermally stable organic units.6–9 As a result, phthalonitrile resins exhibit outstanding thermal-mechanical properties, flame retardancy, chemical resistance, and low moisture absorption, making them attractive for aerospace composites, marine structures, and high-temperature electronic encapsulation.10–15 However, the rigid molecular architecture and strong intermolecular interactions of phthalonitrile moieties result in inherently high melting temperatures (Tm), which impose demanding processing conditions for composite fabrication.16–21
To address the intrinsic processing limitations of phthalonitrile resins, numerous strategies have been explored to depress the Tm of PN. A widely adopted strategy is chemical structure modification. Introducing functional groups such as methyl 22 and epoxy 23 groups to reduce molecular symmetry, incorporating flexible linkages like phosphate and siloxane bridging,24–26 thioether bonds, 27 vinyl 28 or methylene 29 units, or embedding heteroatomic and heterocyclic structures (e.g., fluorinated groups, 30 pyridine rings, 31 or phosphazene 32 bonds) has been demonstrated to reduce the melting temperature of PN. Resins developed through these strategies generally exhibit improved melt processability and enable applicability to resin transfer molding (RTM) processes. However, such structural modification strategies often involve complex synthetic routes, may compromise the intrinsic thermal or mechanical performance, and pose significant challenges for large-scale production.
Another approach to improving PN processability is prepolymerization to generate PN prepolymers. By restricting the degree of polymerization, the prepolymerization of PN monomers and amines can generate low-melting prepolymer with suppressed crystallinity.33,34 Ding 35 prepared PN prepolymer with Tm of approximately 144°C via solution prepolymerization, followed by complicated post-processing steps including filtration, solvent washing, concentration, and drying. Keller 36 reported the melt prepolymerization of PN with p-BAPS and m-APB at 250°C with under vigorous stirring for 10–20 min, producing prepolymer with Tm around 40°C. Solution prepolymerization enables the formation of relatively stable and homogeneous prepolymers; however, residual solvent is difficult to completely remove. Such residues can interfere with curing reactions, reduce crosslinking density, act as plasticizers that increase chain mobility, weaken fiber–matrix interfacial adhesion, and generate internal voids during volatilization, ultimately resulting in diminished thermal stability, mechanical strength, and long-term durability of the composite.37–39 In contrast, melt prepolymerization avoids solvent introduction, but the inherently high melting point of phthalonitrile monomers typically requires reaction temperatures above 200°C, leading to rapid reactions within a narrow time window and limited control over the prepolymerization degree.40,41
In this work, a controllable melt prepolymerization strategy was developed to address the limited controllability of conventional PN melt prepolymerization in PN. By exploiting the eutectic effect among phthalonitrile monomers, the melting and reaction temperature required for melt prepolymerization was significantly reduced, enabling a milder and controllable prepolymerization process to obtain a low-melting phthalonitrile prepolymer MPPh. The evolution of the prepolymerization process was systematically investigated using DSC and FTIR. Subsequently, the prepolymer MPPh was blended with 3-aminophenylacetylene (APA) to prepare MP-m, and the prepolymerization and curing mechanisms were further analyzed by DSC and FTIR. The rheological behavior of the MP-m resin relevant to RTM processing was evaluated through comprehensive rheological analyses. In addition, quartz fiber-reinforced composites (QF/MP-m) were fabricated, and their mechanical properties were investigated. These results highlight the potential of this melt prepolymerization approach for advanced phthalonitrile composite processing, where the incorporation of APA further contributes to enhanced thermal stability and mechanical performance.
Experimental section
Materials
Bisphenol-A, resorcinol, phenol, 4-nitrophthalonitrile and potassium carbonate (K2CO3), 4-nitrophthalonitrile, N,N-Dimethylformamide solvent, acetone solvent and 4,4′- diaminodiphenylmethane (DDM) were obtained from Shanghai Titan Scientific Co., Ltd. (China). 3-aminophenylacetylene (APA) (>98.0%) was supplied by Shanghai Aladdin Bio-Chem Technology Co., Ltd. Quartz fiber fabric (QF) (twill weave, 0.2 mm thickness) was obtained from Hubei Feilihua Quartz Glass Co., Ltd.
Synthesis of 2,2-bis [4-(3,4-dicyanophenoxy) phenyl]propane (BAPh)
BAPh was synthesized according to the following method: To a 250 ml, four-necked round bottom flask was added bisphenol-A (57 g, 0.25 mol), and anhydrous potassium carbonate (75.9 g, 0.55 mol), and 175 g of dry DMF. The mixture was refluxed for 3.5 h at 80–90°C under nitrogen (N2) atmosphere. Subsequently, 4-nitrophthalonitrile (90.8 g, 0.525 mol) was slowly added into the four-necked flask and the compound reacted for an additional 8 h. Lastly, the product mixture was slowly decanted into 300 ml of dilute HCl solution. The solid was collected by suction filtration and washed with water until the filtrate was neutral. The product was filtered and dried under vacuum at 60°C. (Yield: 90%). Purity (HPLC): >98%. FT-IR (KBr, cm-1): 2973 cm−1 (allylic C-H vibration), 1252 cm−1 (aryl ether asymmetric stretching), 2234 cm-1 (C≡N stretching). 1H NMR (600 MHz, DMSO): δ 8.10 (d, J = 8.8 Hz, 2H), 7.79 (d, J = 2.6 Hz, 2H), 7.37 (d, 4H), 7.35 (d, 2H), 7.13 (d, J = 8.7 Hz, 4H), 3.32 (s, 1H), 1.69 (s, 6H). 13C NMR (151 MHz, DMSO): δ 161.05, 151.64, 147.43, 136.32, 128.70, 122.70, 121.87, 119.81, 116.68, 115.92, 115.42, 108.09, 42.13, 30.55. Elemental analysis (%): calculated for C31H20N4O2 (C, 77.49; H, 4.20; N, 11.66; O, 6.66), found (C, 77.26; H, 4.38; N, 11.43; O, 6.93).
Synthesis of 1,3-bis (3,4-dicyanophenoxy) benzene (DPh)
Replacing bisphenol-A with resorcinol as a raw material, the same procedure was repeated for the synthesis of DPh. (Yield: 94.5%). Purity (HPLC): >98%. FTIR (KBr, cm-1): 2234 cm−1 (C≡N stretching), 1246 cm−1 (aryl ether asymmetric stretching). 1H NMR (600 MHz, DMSO): δ 8.13 (d, J = 8.8 Hz, 2H), 7.92 (d, J = 2.6 Hz, 2H), 7.60 (t, J = 8.4 Hz, 1H), 7.55 (dd, J = 8.7, 2.6 Hz, 2H), 7.14 (d, J = 2.6 Hz, 2H), 7.13 (d, J = 2.2 Hz, 1H). 13C NMR (151 MHz, DMSO): δ 160.66, 155.41, 136.29, 132.29, 122.93, 122.66, 117.45, 116.75, 115.39, 112.77, 108.65. Elemental analysis (%): calculated for C22H10N4O2 (C, 72.92; H, 2.78; N, 15.46; O, 8.83), found (C, 72.68; H, 2.95; N, 15.21; O, 9.16).
Synthesis of 1,2-dicyanophenoxy-4-phenoxybenzene (PPh)
PPh was synthesized according to the following method: To a 250 ml, four-necked round bottom flask was added phenol (23.5 g, 0.25 mol), and anhydrous potassium carbonate (37.95 g, 0.275 mol), and 130.6 g of dry DMF. The mixture was refluxed for 3.5 h at 80–90°C under nitrogen (N2) atmosphere. Subsequently, 4-nitrophthalonitrile (45.4 g, 0.2625 mol) was slowly added into the four-necked flask and the compound reacted for an additional 8 h. Lastly, the product mixture was slowly decanted into 300 ml of dilute HCl solution. The solid was collected by suction filtration and washed with water until the filtrate was neutral. The product was filtered and dried under vacuum at 60°C. (Yield: 87.7%). Purity (HPLC): >98%. FTIR (KBr, cm−1): 2234 cm−1 (C≡N stretching), 1253 cm−1 (aryl ether asymmetric stretching). 1H NMR (600 MHz, DMSO): δ 8.10 (d, J = 8.8 Hz, 1H), 7.79 (d, J = 2.6 Hz, 1H), 7.53 – 7.49 (m, 2H), 7.37 (dd, J = 8.8, 2.6 Hz, 1H), 7.33 (t, J = 7.4 Hz, 1H), 7.20 (d, J = 7.6 Hz, 2H). 13C NMR (151 MHz, DMSO): δ 161.05, 153.76, 136.33, 130.67, 125.84, 122.71, 121.93, 120.34, 116.70, 115.91, 115.39, 108.14. Elemental analysis (%): calculated for C14H8N2O (C, 76.35; H, 3.66; N, 12.72; O, 7.26), found (C, 76.18; H, 3.79; N, 12.55; O, 7.48).
Preparation of phthalonitrile prepolymer MPPh
Phthalonitrile prepolymer were prepared in a three-necked flask equipped with a magnetic stir bar, a thermometer and a nitrogen inlet. Upon nitrogen purging for 15 min, the 170°C pre-heated flask was slowly charged with PPh (50 g), DPh (33 g), and BAPh (17 g) under stirring. Once ternary phthalonitrile monomer mixture completely melted, DDM (2 g) were slowly added into the flask. After stirring at 170°C for 3 h, the prepolymer were poured out and quenched to room temperature, yielding the low-melting phthalonitrile prepolymer MPPh.
Preparation of MP-m
Predetermined amounts of MPPh resin and APA (corresponding to 5 wt%, 10 wt%, and 15 wt% of the MPPh mass, respectively) were weighed and placed into a three-necked flask. Under a nitrogen atmosphere, the mixture was heated to 60°C and maintained in the molten state with continuous stirring for 0.5 h. The resulting blend was then poured out while still hot and allowed to cool to room temperature, yielding the MP-m5, MP-m10, and MP-m15 resins.
Preparation of MPPh and MP-m thermosets
The MPPh, MP-m5, MP-m10, and MP-m15 resins were cured in a convection oven according to the following temperature profile: 220°C/1 h, 260°C/2 h, 300°C/2 h, 350°C/5 h, with a constant heating rate of 5°C/min. Upon completion of the curing cycle, the samples were cooled to room temperature under ambient conditions and subsequently removed, resulting in black cured materials.
Preparation of QF/MPPh and QF/MP-m composites
Preparation of QF/MPPh and QF/MP-m prepreg by solution impregnation method: MPPh and MP-m resin was dissolved in acetone to prepare a homogeneous resin solution, into which quartz fiber fabric (QF) was uniformly immersed. The mass ratio of MPPh and MP-m resin to fiber was maintained at 35:65. The impregnated fabric was then placed in a fume hood for 12 h to allow acetone evaporation, yielding the QF/MPPh and QF/MP-m prepreg.
Molding of QF/MPPh and QF/MP-m composites by compression molding: Fourteen layers of the prepared prepregs were cut to dimensions of 8 cm × 20 cm and stacked with identical warp and weft orientations before being placed in a flat-plate vulcanizing press for compression molding (CMG50H-18-PX). A molding pressure of 6 MPa was applied, and the curing process followed the same procedure as previously described. After cooling and demolding, the QF/MPPh and QF/MP-m composites were obtained.
Characterization
High-performance liquid chromatography (HPLC) was performed using a Vanquish UHPLC system (Thermo Fisher, Germany) with methanol as the eluting solvent. 1H NMR and 13C NMR spectra were collected on a Bruker AVANCE 500 spectrometer (Bruker, Germany) at room temperature using DMSO as the solvent and internal standard. Elemental analysis was conducted with a Vario EL Cube Analyzer (Elementar, Germany), with precision of C, H, N, O ≤0.3%. Differential Scanning Calorimetry (DSC) analysis was performed using a DSC 200 F3 from NETZSCH, Germany, to investigate the heat absorption and exothermic curves of monomers, mixtures D, prepolymers MPPh and MP-m resins during the curing process, within the temperature range of 50–400°C. The viscosity of sampled MPPh during melt prepolymerization process were measured using a parallel plate viscometer (model CAP2000+H, Brookfield Engineering Laboratories, USA), with the measurement conditions (50°C, 20 rpm, 1 mm gap). The rheological characteristic of MPPh and MP-m was studied by a Discovery H-1 rheometer from TA Instruments, USA, at a strain of 0.02 N with a frequency of 1 Hz. The rheological characteristic of MPPh and MP-m were studied by Discovery H-1 rheometer Discovery H-1 rheometer from TA Instruments, USA, with a testing temperature range of 60–260°C at a strain of 0.02 N with a frequency of 1 Hz. The prepolymerization of MPPh and curing process of MPPh and MP-m resins was further analyzed using a Nicolet 6700 Fourier Transform Infrared Spectrometer (FTIR) from Thermo Nicolet Corpo ration, USA, over the wavelength range of 4000–400 cm−1. The curing process of MPPh and MP-m resins in air and nitrogen was also examined through mass loss tests using a TGA 209F3 thermogravimetric analyzer (TGA) from NETZSCH, Germany, with temperature ranges of 50–1000°C. The mechanical properties of QF/MPPh and QF/MP-m composites were evaluated using an E44/304 universal tensile testing machine from China Meters Industrial Systems Co. The tensile properties were determined according to GB/T1447-2005, the bending properties according to GB/T1449-2005, and the interlaminar shear properties following GB/T30969-2014. The dynamic thermomechanical properties of QF/MPPh and QF/MP-m composites were analyzed using a Q800 Dynamic Mechanical and Thermal Analyzer (DMA) from TA Instruments, USA, with a testing temperature range of 50–400°C. The surface morphology of QF/MPPh and QF/MP-m composites before and after thermal aging, was investigated using an S-4800 Field Emission Scanning Electron Microscope (SEM) from Hitachi, Japan. The QF/MPPh and QF/MP-m composites were prepared using a CMG50H-18-PX flat-plate vulcanizing press from Caever, USA.
Result and discussion
Eutectic-enabled melt prepolymerization of MPPh
To reduce the temperature required for melt prepolymerization and improve its controllability, a eutectic blending approach was investigated. As shown in Figure 1(a), the melting temperatures (Tm) of the synthesized phthalonitrile monomers BAPh, DPh, and PPh are 197.5°C, 186.3°C, and 101.6°C, respectively. The single melting peak suggests that all monomers are crystalline. The ternary mixture was analyzed by DSC, as shown in Figure 1(b). An endothermic peak at 99.5°C is assigned to the eutectic temperature (Te), which is lower than either pure component suggesting a eutectic composition.
42
In eutectic mixtures, intimate contact between dissimilar crystalline species promotes lattice penetration and bond rearrangement at crystal interfaces, which amplifies atomic vibrational instability (Lindemann criterion) and initiates surface-driven melting at temperatures far below the melting points of the individual components.
43
In addition, a broad endothermic peak centered at approximately 123.6°C is observed and assigned to the Tm of the ternary mixture, which is characteristic of eutectic systems.
44
Even thought, after heating to 150°C the ternary monomer mixture can form a homogeneous brownish-yellow solution, suitable for melt prepolymerization as present in Figure 2(a). DSC curves of BAPh, DPh, and PPh (a) and their ternary mixture (b) with a mass ratio of BAPh:DPh:PPh = 1.7:3.3:5. Tm and Te indicate melting and eutectic temperatures. (a) Digital images of the processes of monomer mixture with a mass ratio of BAPh:DPh:PPh = 1.7:3.3:5 and melt prepolymerization. (b) Digital images of sampling and solubility at different reaction times during melt prepolymerization. (c) Viscosity of different reaction time during melt prepolymerization.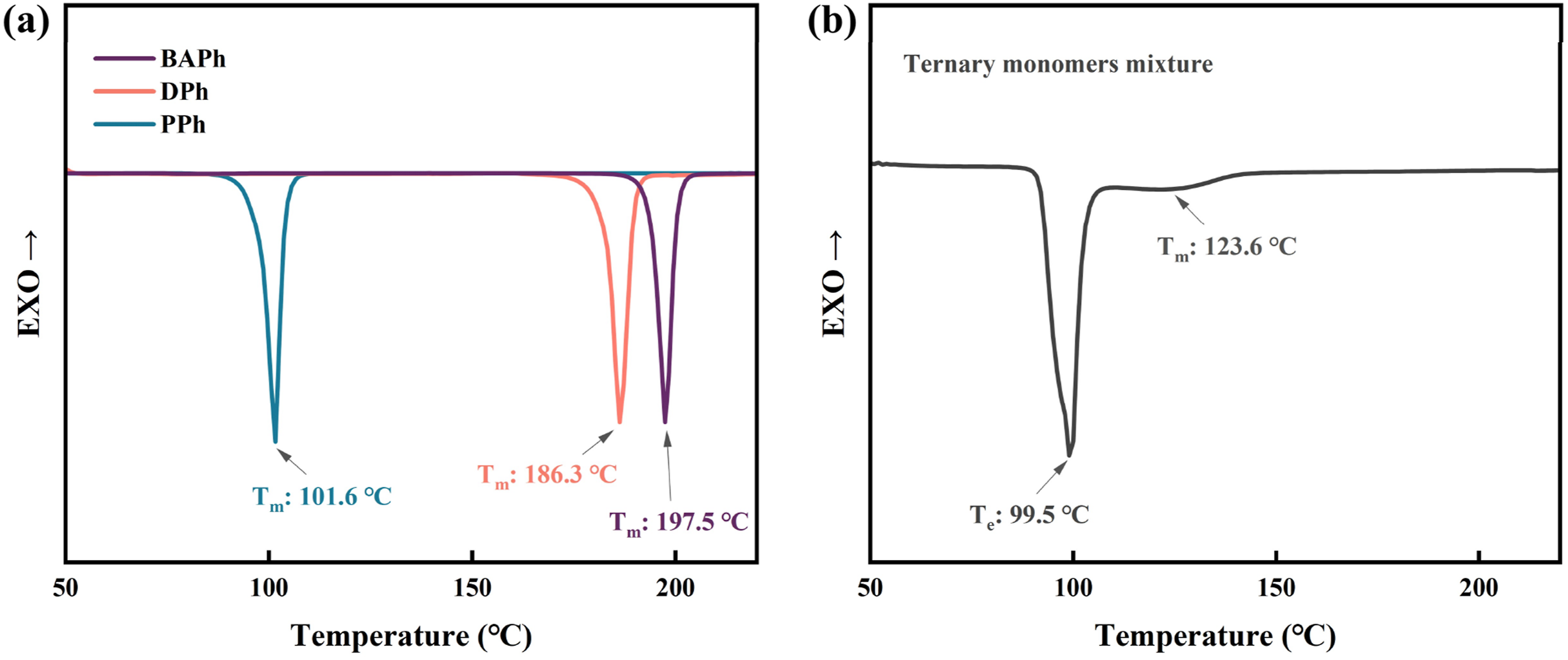
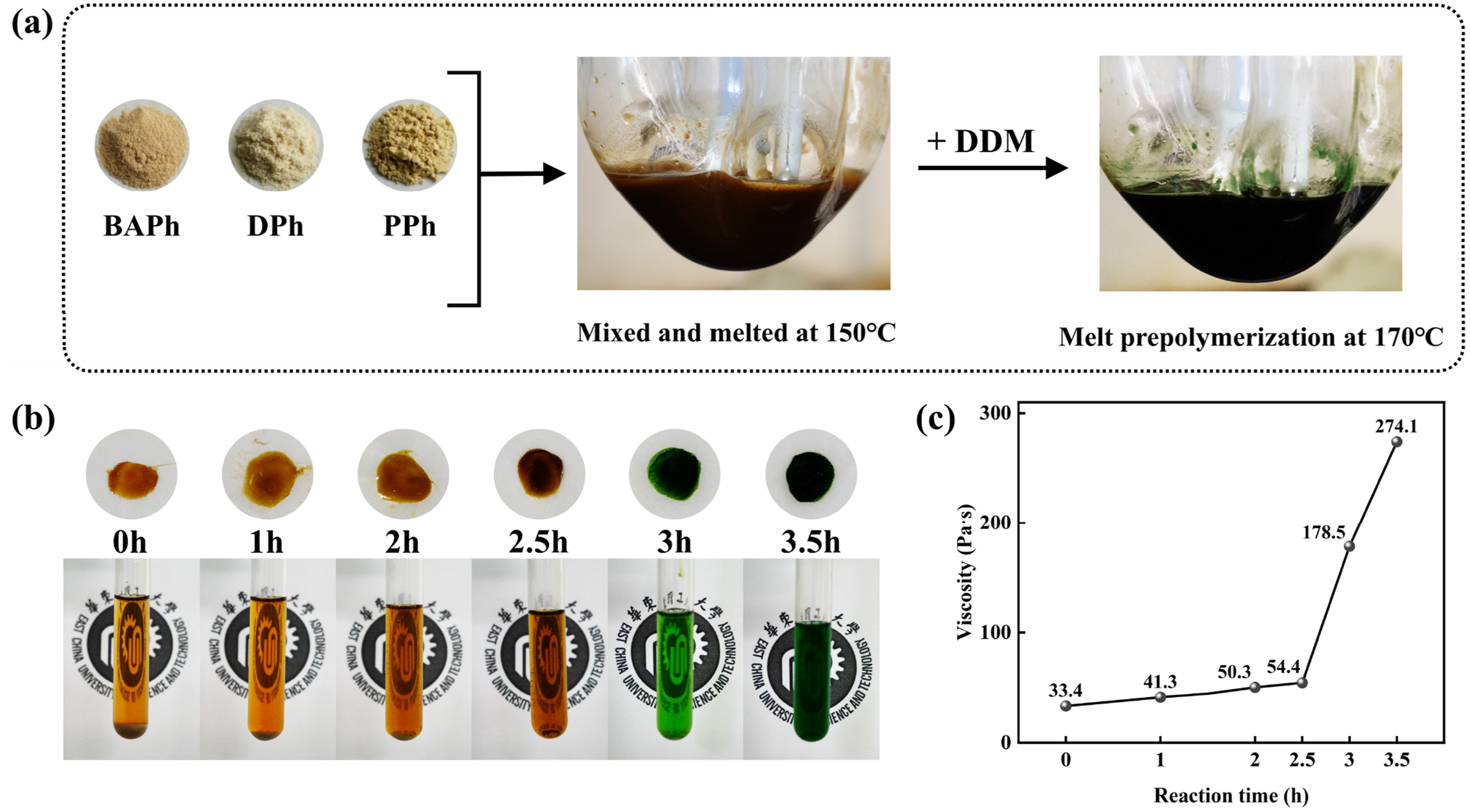
A low-melting, amorphous phthalonitrile prepolymer (MPPh) was prepared via melt prepolymerization of the ternary monomer mixture in the presence of 2 wt% DDM. After adding DDM and conducting the melt prepolymerization reaction for 3.5 h, the color turned dark green. To further monitor the prepolymerization process, the prepolymers were extracted at a reaction time ranging from 0 to 3.5 h, and its state, solubility and viscosity were observed as presented in Figure 2(b) and (c). During the reaction process, the color changed from brownish yellow to green, and this change occurred from 2.5 h to 3 h. The solubility of the prepolymers were evaluated by dissolving the samples in acetone at a solid content of 40 wt% at room temperature. The poor solubility of samples sampled at the beginning of the reaction was attributed to incompletely reacted BAPh and DPh monomers. After 2.5 h, the solubility significantly improved, indicating phthalonitrile monomers were involved in the reaction. Meanwhile, the viscosity of prepolymers increased gradually during the first 2.5 h, reaching 54.4 Pa·s, which suggests a moderate and controllable reaction in the early stage of prepolymerization. In contrast, a pronounced increase in viscosity to 178.5 Pa·s was observed when the reaction time was extended to 3 h. The pronounced changes in solubility, color, and viscosity between 2.5 h and 3 h provided compelling macroscopic evidence of melt prepolymerization, indicating that the significant chemical structure evolution occurred within this interval and warranting further analysis.
To elucidate the prepolymerization mechanism, prepolymers collected at different reaction times were further analyzed by DSC and FTIR. As shown in Figure 3(a), the prepolymers obtained within 0–2.5 h exhibited an endotherm at approximately 125°C, corresponding to the Tm, and a pronounced exothermic curing peak at 224–252°C, assigned to the curing peak temperature (Tp). The melting endotherm indicates the presence of unreacted monomers or low-molecular-weight oligomers, consistent with the solubility behavior at this stage of prepolymerization. The exothermic peak is characteristic of the amine-catalyzed cyclotrimerization of nitrile groups.
45
After 3 h of reaction, both the endotherm and sharp curing exotherm peak disappear, indicating the formation of an amorphous oligomeric structure and the substantial consumption of reactive amine species during prepolymerization. The evolution of chemical structures during melt prepolymerization was further examined by FTIR (Figure 3(b) and (c)). The absorption band corresponding to nitrile groups (2232 cm−1, −C≡N) gradually decreases in intensity over time, indicating partial participation of nitrile groups in the reaction. A pronounced spectral change occurs after approximately 2.5 h, where the absorption bands of amino groups (3469 cm-1 and 3378 cm−1, −NH2) decrease concurrently with a significant increase in the C = N absorption at 1660 cm−1. This change indicates that nucleophilic addition between amino and nitrile groups dominates the prepolymerization process, leading primarily to linear chain growth.
46
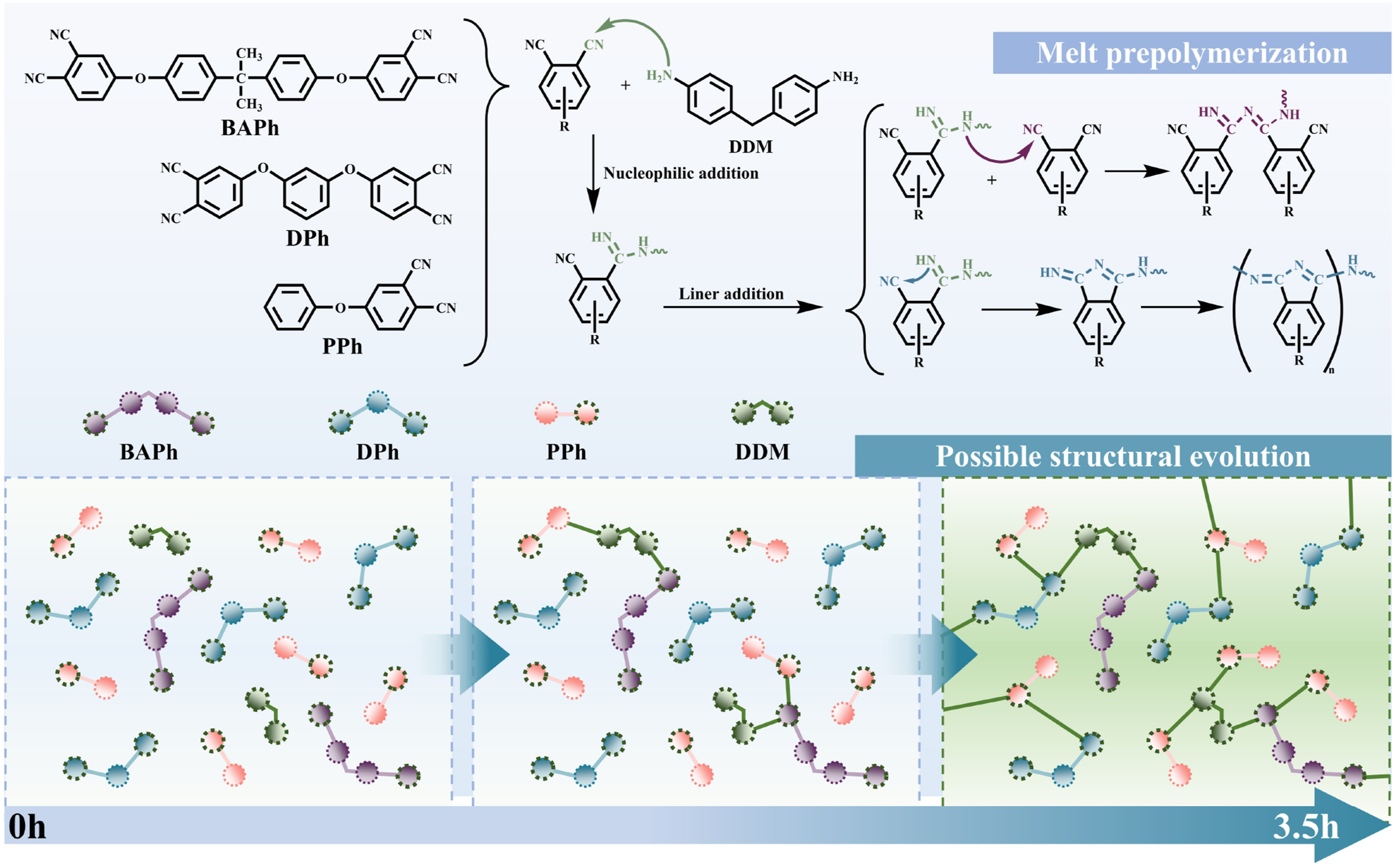
In contrast, no noticeable increase was observed in the characteristic absorption bands associated with triazine structures (1475 cm−1), isoindoline structures (1725 cm−1), or phthalocyanine ring structures (1010 cm−1) even after 3.5 h, which are typically formed through extensive cyclization and crosslinking of nitrile groups.47,48 This observation indicates that the prepolymerization primarily involves the consumption of amino and nitrile groups through addition reactions, leading to linear chain growth rather than significant ring-forming polymerization. Accordingly, a possible reaction path for the melt prepolymerization of ternary monomer mixture and DDM is shown in Figure 4. Combined with the phenomenon that the monomers fully involved to react to form the prepolymer and the color turned green and the viscosity increased rapidly when the reaction reaches 3 h, this reaction time was determined as the end point of the melting prepolymerization reaction to obtain MPPh. (a) DSC curves of MPPh during melt prepolymerization at different reaction times. (b) FTIR spectra of MPPh during melt prepolymerization at different reaction times. (c) FTIR spectra of the main characteristic absorption bands at different reaction times during melt prepolymerization of MPPh. Schematic illustration of melt prepolymerization mechanism of MPPh.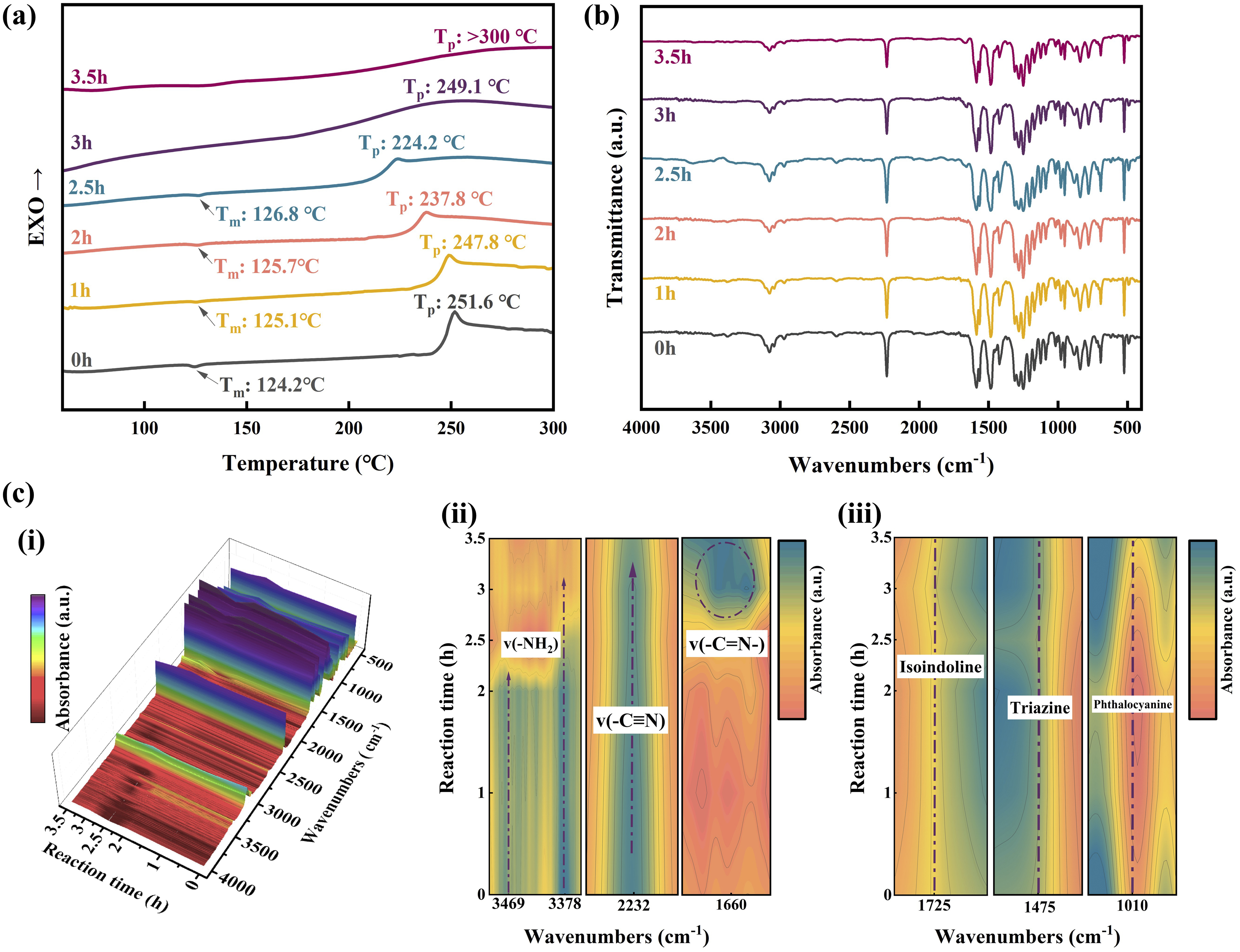
Curing reactivity and rheology of MPPh and MP-m
The curing reactivity of MPPh and MP-m was analyzed by DSC, gel time and complex viscosity-temperature as shown in Figure 5. MPPh exhibited no distinct exothermic peak and reaction enthalpy (ΔH) was only 115.7 J g−1, and gel time exceeded 100 min at 240°C. Although viscosity-temperature curve indicated that the viscosity of MPPh could be reduced to 1 Pa·s at 88°C, no significant viscosity increase was observed even when the temperature reached 250°C. This insufficient curing reactivity severely limits the practical processability of MPPh in composite manufacturing. (a) DSC curves of MPPh, MP-m5, MP-m10, and MP-m15 resins. (b) Gel time of MPPh, MP-m5, MP-m10, and MP-m15 resins. (c) Viscosity versus temperature curves of MPPh, MP-m5, MP-m10, and MP-m15 resins.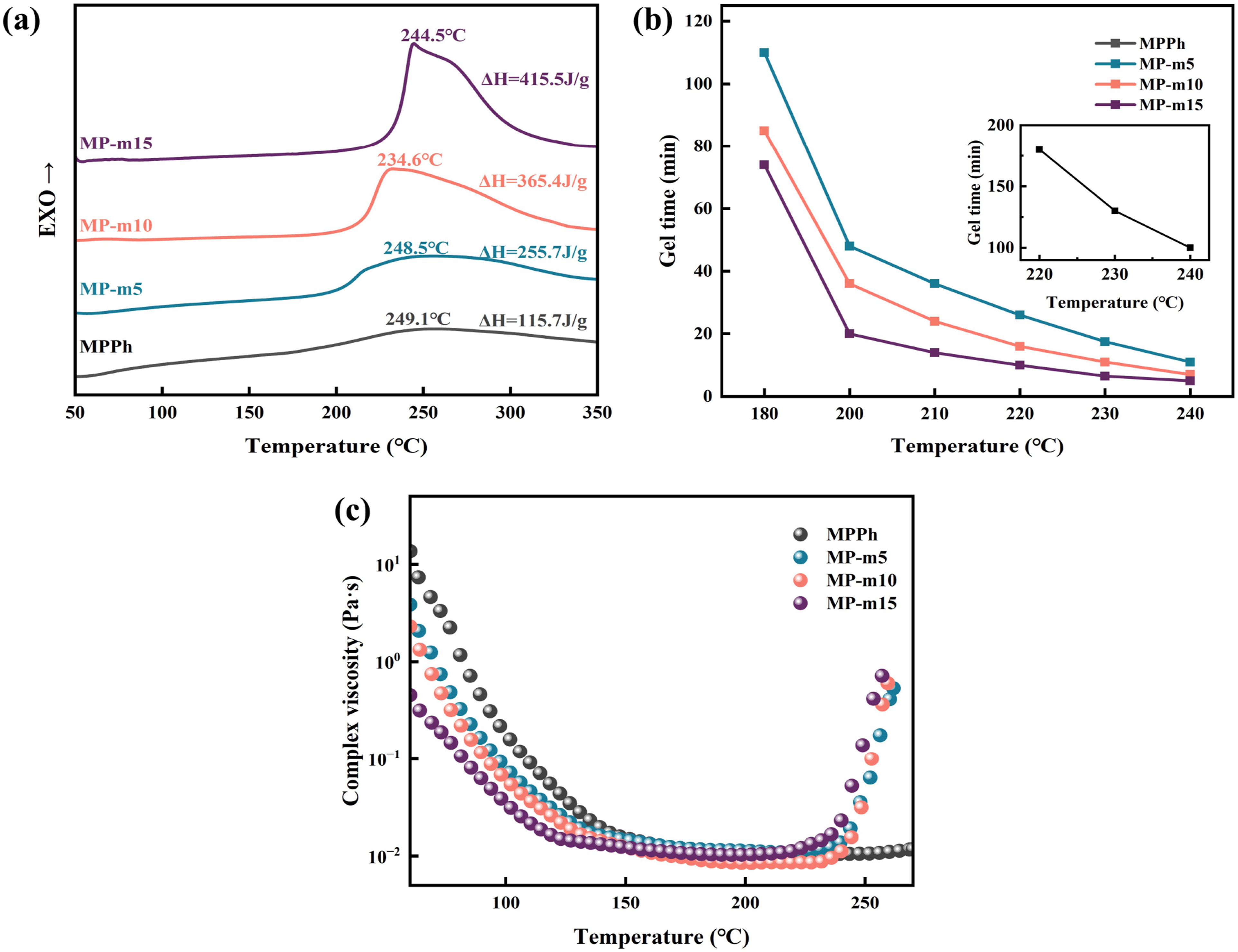
To overcome this limitation, 3-aminophenylacetylene (APA) was incorporated into MPPh to obtain the modified resins MP-m5, MP-m10, and MP-m15. With increasing APA content, systematic changes in curing behavior were enhanced. As shown in Figure 5(a), all MP-m resins exhibit a single dominant exothermic peak centered at approximately 240°C, attributed to the amino-catalyzed polymerization of nitrile groups. The curing onset temperature increases progressively from 206°C for MP-m5 to 235°C for MP-m15 due to reduced intermolecular interactions and increased molecular mobility induced by APA incorporation, which delays the initial thermal activation of the curing reaction. Meanwhile, the total curing enthalpy increased markedly with APA content, rising from 255.7 J g−1 for MP-m5 to 365.4 J g−1 for MP-m10 and further to 415.5 J g−1 for MP-m15. The higher curing enthalpy indicated a greater extent of crosslinking and a more complete curing reaction in the MP-m. The gel time (Figure 5(b)) of MP-m was significantly shortened after the addition of APA, decreasing to below 30 min at 220°C. Meanwhile, Rheological measurements (Figure 5(c)) further showed that MP-m presented a significant increase in viscosity around 250°C compared with MPPh, consistent with the results of DSC and gel time. In addition, the incorporation of APA improved the rheological behavior. The MP-m resins exhibited a minimum melt viscosity of 0.01 Pa·s at approximately 200°C. Notably, MP-m10 reached a viscosity of 1 Pa·s at 66°C and maintained a practical processing window of 66–92°C, within which the melt viscosity ranges between 0.1 and 1 Pa·s24. This reduction in melt viscosity is attributed to the small molecular size of APA, which penetrates between prepolymer chains, weakens intermolecular interactions, and thereby facilitate molecular mobility.
Furthermore, viscosity-time measurements of MP-m10 were performed at 80°C, 110°C, and 140°C, as shown in Figure 6(a). The viscosity remained stable at approximately 0.47, 0.06, and 0.03 Pa·s, respectively, with no detectable increase over time. These temperatures lie well within the processing window and far below the curing temperature, indicating that MP-m10 maintains a stable, low viscosity under prolonged thermal exposure, which is advantageous for processes requiring extended mold filling and pressure-holding times. Figure 6(b) presented the viscosity-shear rate behavior of MP-m10 over a shear rate range of 0.1–100 s−1 at 80°C, 110°C, and 140°C. The viscosity slightly decreased with increasing shear rate, indicating typical shear-thinning, non-Newtonian behavior. This shear-thinning characteristic is particularly beneficial for RTM,
49
as it facilitates resin infiltration under shear while maintaining low viscosity during mold filling, thereby enabling the fabrication of complex composite structures. (a) Viscosity versus time curves of MP-m10 resin at 80°C, 110°C and 140°C. (b) Viscosity versus shear rate curves of MP-m10 resin at 80°C, 110°C and 140°C.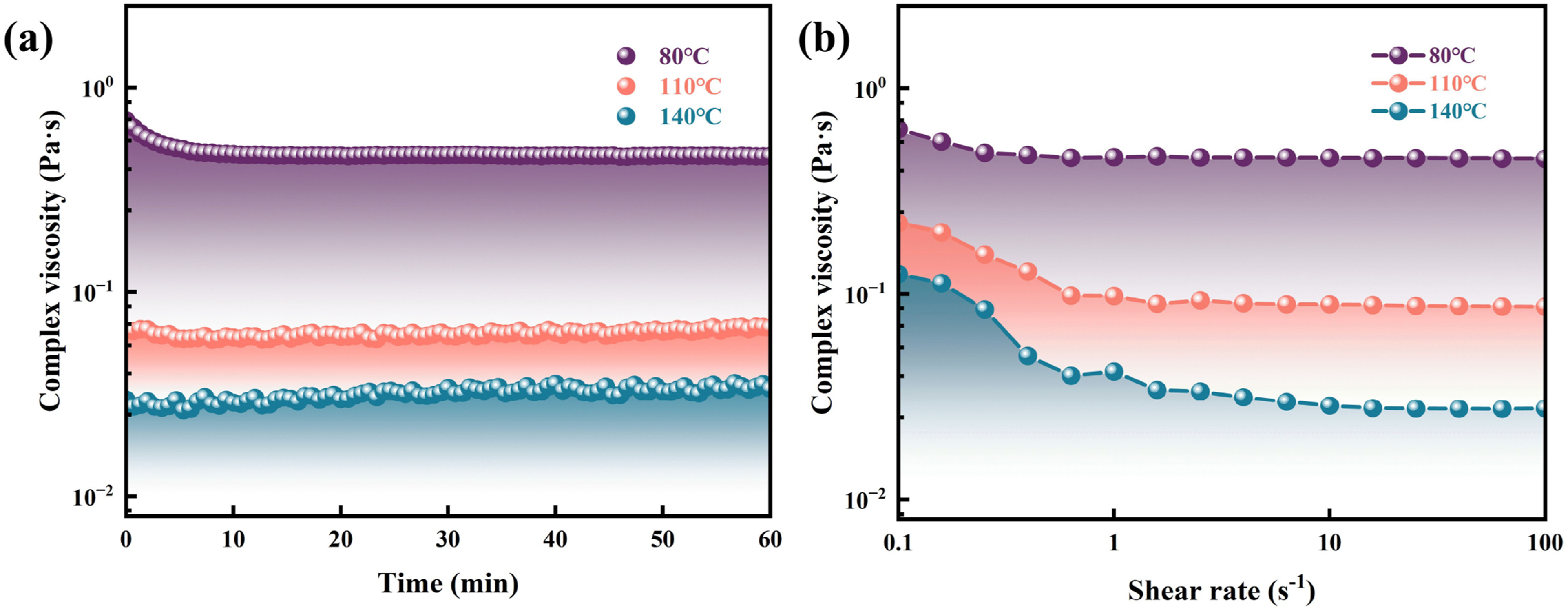
The curing behavior of MPPh and MP-m resins was investigated by FTIR, as shown in Figure 7. In the spectra of the uncured MPPh and MP-m resins (Figure 7(a)), a distinct absorption band at 2223 cm−1 corresponding to nitrile groups (–C≡N) was observed. The absorption near 3030 cm−1 is attributed to the aromatic C–H stretching vibration. In contrast, the FTIR spectrum of APA displays characteristic bands of terminal alkynyl groups at approximately 3284 cm−1 and a weak band near 2204 cm−1 assigned to C≡C stretching, together with amino group (–NH2) absorptions at 3471 cm−1 and 3361 cm−1. After incorporating APA into MPPh, the uncured MP-m samples clearly exhibited the characteristic amino and alkynyl absorption bands originating from APA, while no additional new peaks or significant peak shifts were detected. This observation indicates that APA is physically dispersed within the prepolymer matrix without undergoing covalent reactions prior to thermal curing. (a) FTIR spectra of uncured MPPh, MP-m5, MP-m10, and MP-m15 resins. (b) FTIR spectra of MPPh, MP-m5, MP-m10, and MP-m15 resins were cured at a maximum of 350°C. (c) FTIR spectra of MP-m10 resins were cured at a maximum of 170°C, 260°C, and 350°C, respectively.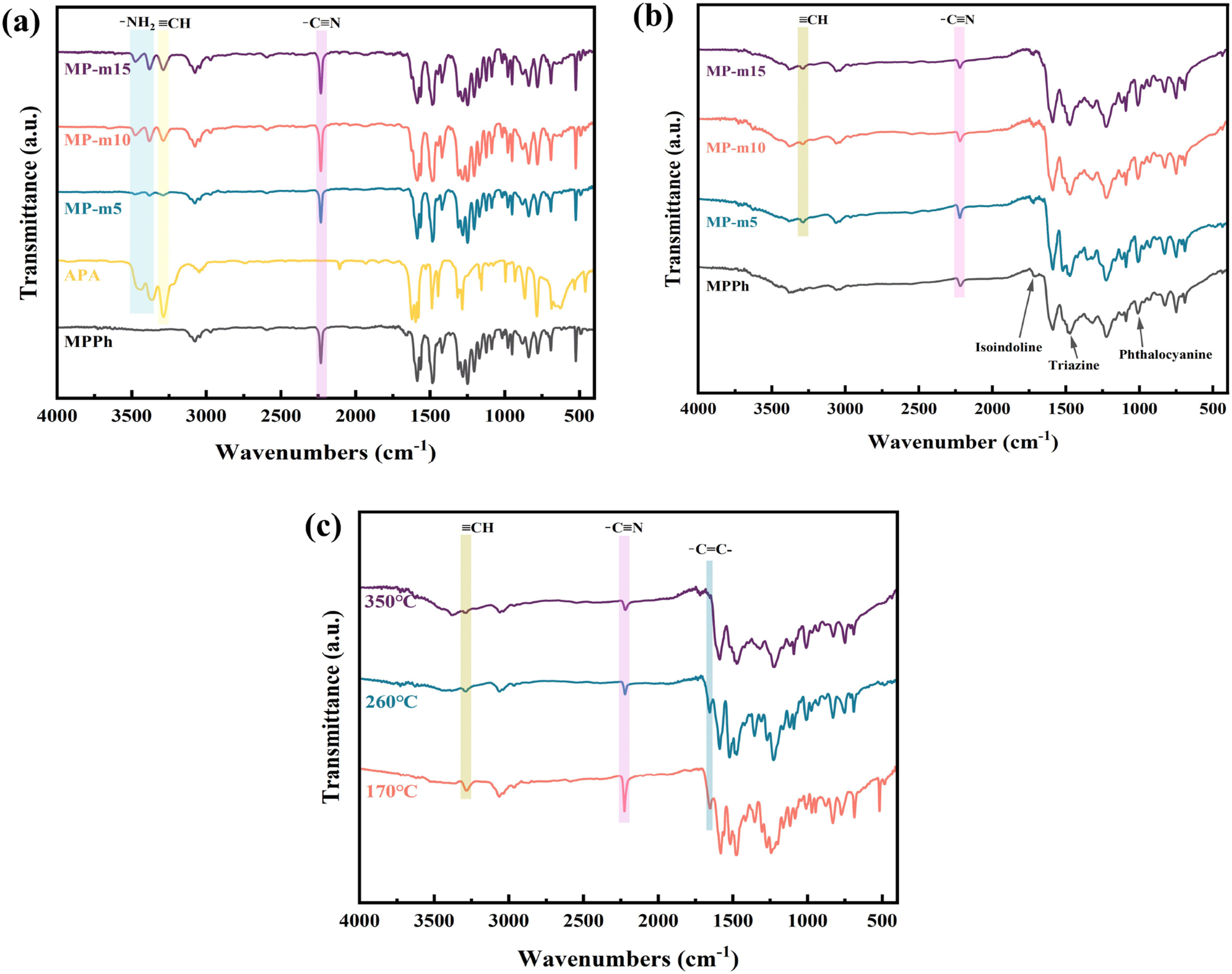
After curing at 350°C (Figure 7(b)), substantial spectral changes are observed for both MPPh and MP-m. The intensities of the –C≡N, –NH2, and H–C≡C– absorption bands decrease markedly, indicating extensive consumption of nitrile, amino, and alkynyl groups during curing. Meanwhile, several new characteristic bands emerge, associated with the formation of a highly crosslinked network. The band at approximately 1475 cm−1 is attributed to triazine structures, the absorption near 1725 cm−1 corresponds to isoindoline structures, and the band around 1010 cm−1 is assigned to phthalocyanine ring structures, consistent with previously reported curing mechanisms of phthalonitrile resins.46,50–52 Compared with MPPh, the APA-modified MP-m resins exhibited more pronounced consumption of reactive functional groups and stronger absorptions associated with cured network structures, indicating that APA promotes curing reactions and enhances network formation. These observations are consistent with the DSC and gelation results.
Figure 7(c) presents the FTIR spectra of MP-m10 cured at 170°C, 260°C, and 350°C, providing insight into the structural evolution during curing. As the curing temperature increased, the absorption bands of terminal alkynyl groups at 3288 cm−1 and nitrile groups at 2231 cm−1 gradually decrease in intensity, indicating their participation in the curing reactions. A new absorption band at 1651 cm−1 (–C = C–) appeared after curing at 170°C and becomes nearly absent at 350°C, suggested that the H–C≡C– groups in APA undergo Diels–Alder reactions in the range of 170–260°C to form polyene structures, which subsequently participate in further crosslinking at higher temperatures.53,54 Considering the relatively high content of APA introduced as a reactive diluent, it is reasonable to infer that, in addition to co-curing with nitrile groups, some alkynyl groups may also undergo self-reactions at elevated temperatures, generating substituted aromatic structures that further densify the network. Meanwhile, the absorption bands associated with isoindoline, triazine, and phthalocyanine ring structures progressively intensify as the curing temperature increases from 170°C to 350°C. In addition, the nitrile absorption band shifts toward lower wavenumbers during curing, further supporting the formation of triazine derivatives. 55 The progressive enhancement of these characteristic bands indicates the development of a highly crosslinked network dominated by thermally stable heterocyclic structures.
Thermal properties of MPPh and MP-m
The thermal performance of cured MPPh and MP-m resins was evaluated by thermogravimetric analysis (TGA). Figure 8(a) and (c) presented the TGA curves under nitrogen and air atmospheres, respectively, and Table. 1 summarized the corresponding thermal decomposition parameters, including the temperature at 5% weight loss (Td5), the temperature at the maximum-weight loss rate (Tmax), and the residual char yield at 800°C (Cy800). Compared with MPPh, the cured MP-m resins exhibit improved thermal stability under nitrogen. In particular, MP-m10 shows a Td5 of 515.7°C and a Cy800 of 76.9%. Similarly, under air, the MP-m resins demonstrate enhanced thermo-oxidative stability, with MP-m10 reaching a Td5 of 502.7°C and a Cy800 of 48.6%. TGA and DTG curves of the cured MPPh, MP-m5, MP-m10, and MP-m15 resins under nitrogen (a) and (b) and air (c) and (d) atmospheres. TGA results of the cured MPPh, MP-m5, MP-m10, and MP-m15 resins under nitrogen and air atmospheres.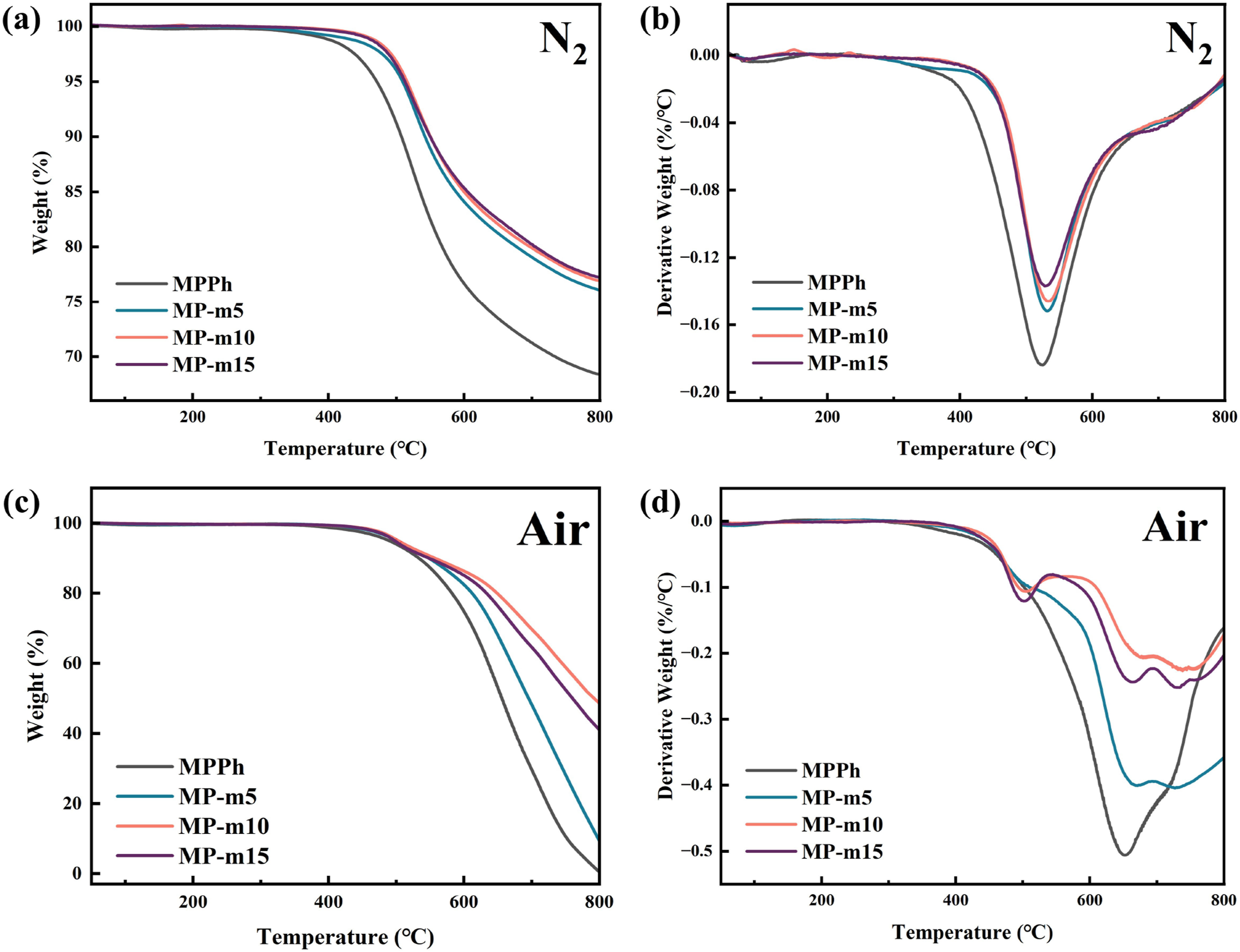
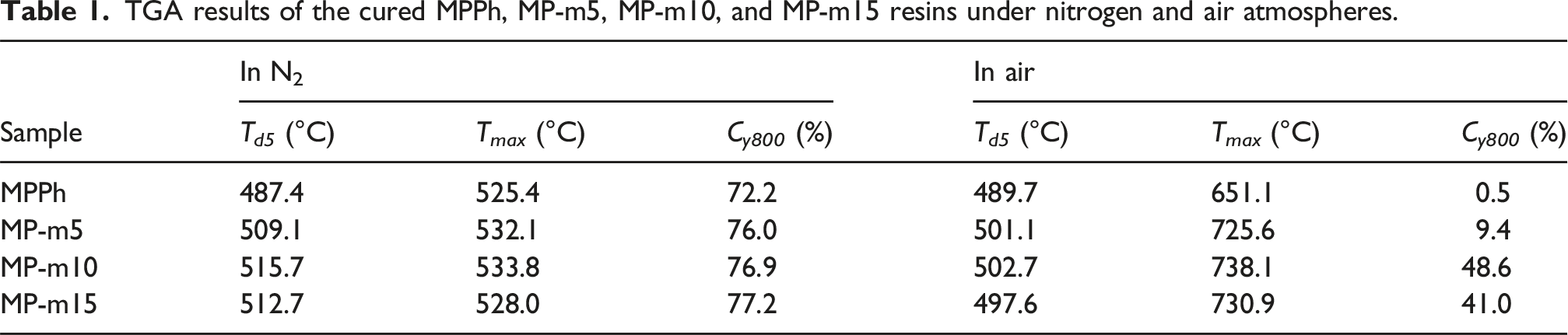
With increasing APA content, both thermal and thermo-oxidative stability initially increase and then decrease, indicating that an appropriate APA content promotes nitrile curing and increases the crosslink density. The subsequent decline in stability at higher APA contents is attributed to the formation of trisubstituted benzene and polyene-type structures with relatively lower thermal stability resulting from APA self-polymerization.
The thermal degradation behavior was further analyzed using derivative thermogravimetric (DTG) curves under nitrogen and air, as shown in Figure 8(b) and (d). Under nitrogen (Figure 8(b)), the initial decomposition temperature of MPPh occurs at approximately 380°C, whereas that of MP-m resins shifts to around 400°C. This shift indicates the formation of a more densely crosslinked network in the presence of APA. Both MPPh and MP-m exhibit a major decomposition peak near 530°C, which is associated with the cleavage of highly stable heterocyclic structures such as triazine and phthalocyanine formed during curing. 56 The similar peak temperatures suggest that the fundamental high-temperature degradation mechanism remains unchanged after APA incorporation. In contrast, the DTG curves obtained in air (Figure 8(d)) reveal distinct differences in thermo-oxidative degradation behavior. MPPh shows a single dominant weight-loss peak centered at approximately 651°C, corresponding to the oxidative degradation of stable heterocyclic structures. However, MP-m resins exhibit two weight-loss peaks at around 500°C and 670°C. The high-temperature peak is attributed to oxidative decomposition of triazine and phthalocyanine rings, while the additional lower-temperature peak is associated with APA-derived structures possessing lower thermo-oxidative stability. 57 These results indicate that APA enhances the overall network stability while introducing an additional oxidative degradation pathway.
Mechanical properties of MPPh and MP-m composites
Figure 9 shows the mechanical properties of QF/MPPh and QF/MP-m composites. It can be seen that the APA-modified QF/MP-m composites exhibit strong mechanical properties compared to the QF/MPPh composites, and the flexural strength of the QF/MP-m10 composites is 660.1 MPa, which is an increase of 26.0%, the flexural modulus is 32.9 GPa, which is an increase of 12.7%, the interlaminar shear strength is 69.7 MPa, which is an increase of 54.2%, and the tensile strength is 660.7 MPa, which is an increase of 62.8%. (a) Flexural strength and flexural modulus of QF/MPPh, QF/MP-m composites. (b) Tensile strength and interlaminar shear strengths of QF/MPPh, QF/MP-m composites.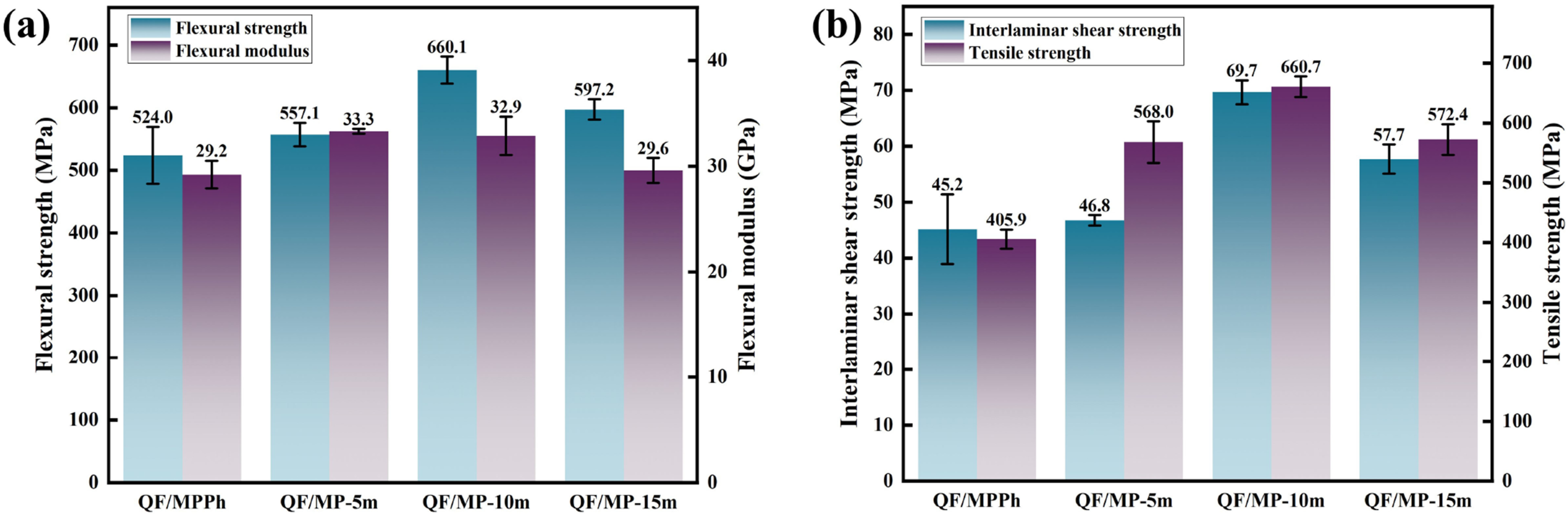
The mechanical performance of the composites is closely related to the interfacial adhesion between the resin matrix and the quartz fibers. The flexural fracture surfaces of QF/MPPh and QF/MP-m composites are shown in Figure 10(a)–(d). In the QF/MP-m10 and QF/MP-m15 composites, the resin remains tightly adhered to the fiber surfaces after fracture, indicating effective impregnation and wetting during fabrication. In combination with the DSC and gelation results, this observation suggests that APA promotes more uniform and complete curing, thereby reducing stress concentrations associated with uneven crosslinking. As a result, strong interfacial bonding is established, enabling efficient load transfer from the resin matrix to the fibers and contributing to improved flexural strength. The interlaminar shear fracture surfaces of QF/MPPh and QF/MP-m composites are presented in Figure 10(a)–(d). In all composites, the gaps between fibers are well filled with resin, indicating good interfacial contact. However, for QF/MPPh, the fracture surfaces of both the quartz fibers and the surrounding resin appear relatively smooth, indicating limited interfacial adhesion and brittle fracture behavior. In contrast, the fracture surfaces of the QF/MP-m composites are noticeably rougher, suggesting enhanced interfacial interactions and improved fracture resistance. This improvement is attributed to the polar functional groups in APA (–NH2), which can interact with hydroxyl groups on the fiber surface through hydrogen bonding, thereby strengthening interfacial adhesion and enhancing interlaminar shear strength.
58
A clear difference in fracture characteristics is observed between QF/MP-m10 and QF/MP-m15. While QF/MP-m15 still exhibits relatively brittle fracture features, QF/MP-m10 shows a tortuous and branched crack propagation path, indicative of improved toughness. In addition, the tensile strength of the QF/MP-m composites increases after modification. The enhanced interfacial adhesion facilitates effective stress transfer under tensile loading, reducing premature fiber debonding and contributing to improved tensile performance. SEM images of flexural failure surface (a, b, c, d) of QF/MPPh and QF/MP-m; SEM images of interlaminar shear failure surface (a’, b’, c’, d’) of QF/MPPh and QF/MP-m.
Dynamic mechanical analysis (DMA) was performed on QF/MPPh and QF/MP-m10 composites, and the storage modulus (E′) and loss factor (tan δ) curves are presented in Figure 11. The storage modulus of both composites remains relatively stable over the measured temperature range. No distinct tan δ peak is observed below 400°C, indicating a high degree of curing and suggesting that the glass transition temperature exceeds 400°C, consistent with the excellent thermal resistance of phthalonitrile networks.59,60 At 50°C, the storage modulus of QF/MPPh and QF/MP-m10 is 11,211 MPa and 19,036 MPa, respectively. The higher storage modulus of QF/MP-m10 is attributed to the incorporation of APA, which increases the effective crosslink density and restricts molecular chain mobility through the participation of its amino and alkynyl groups in the curing reaction. DMA curves of QF/MPPh (a) and QF/MP-m10 (b) composites.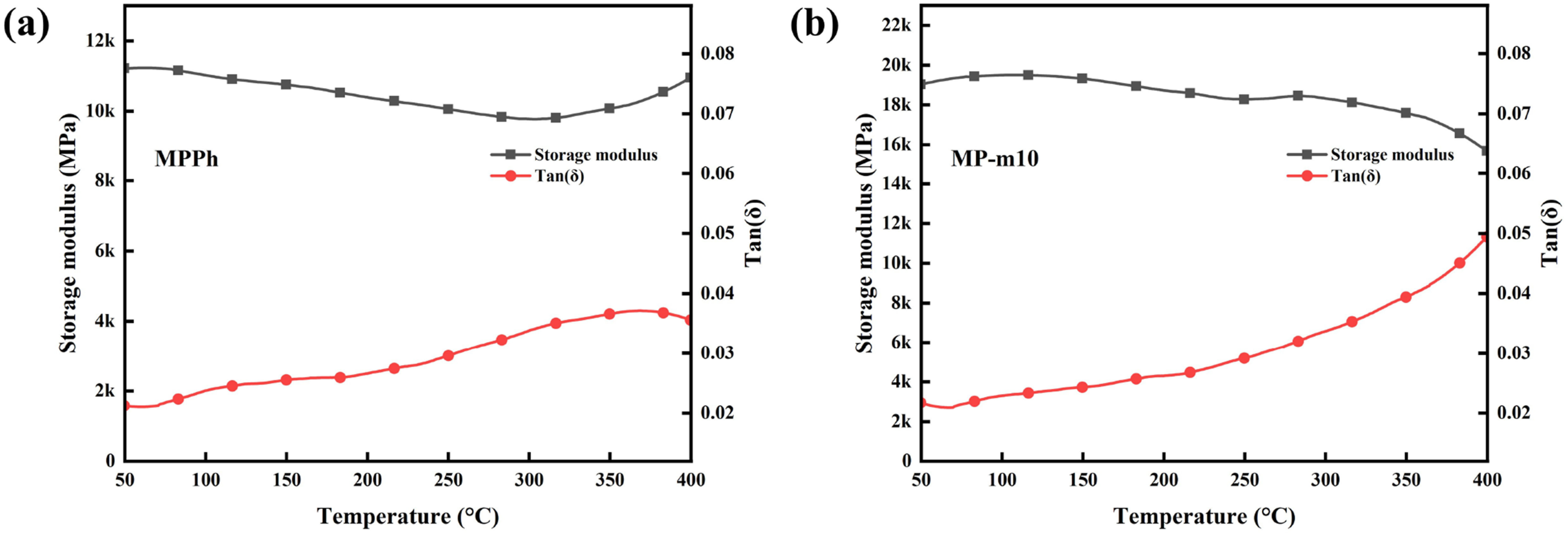
Conclusion
In this work, a controllable melt prepolymerization strategy was established to prepare a low-melting phthalonitrile prepolymer (MPPh) with improved processability. By employing a ternary phthalonitrile monomer blend, the melting temperature was significantly reduced through a eutectic effect, enabling melt prepolymerization at lower temperatures and avoiding the premature gelation typical of conventional high-melting phthalonitrile systems. Combined viscosity, DSC, and FTIR analyses indicate that the prepolymerization proceeds predominantly through linear chain growth via reaction between amino and nitrile groups, resulting in an amorphous oligomeric prepolymer with low melt viscosity.
The incorporation of 3-aminophenylacetylene (APA) further tailored the curing behavior and processing characteristics of MPPh. APA reduced the melt viscosity, shortened gelation time, and broadened the processing window while maintaining controllable curing characteristics. The modified resins (MP-m) exhibited stable viscosity under isothermal conditions and pronounced shear-thinning behavior, demonstrating suitability for liquid composite molding processes. After curing, MP-m10 showed high thermal and thermo-oxidative stability (Td5 = 515.7°C in N2; 502.7°C in air). The QF/MP-m10 composites achieved flexural, interlaminar shear, and tensile strengths of 660.1 MPa, 69.7 MPa, and 660.7 MPa, respectively, representing significant improvements over QF/MPPh composites due to enhanced resin impregnation and optimized curing behavior. This study provides a practical and scalable approach that integrates eutectic-assisted melt prepolymerization with reactive modification to produce phthalonitrile resins with excellent melt processing properties without loss of thermal stability and mechanical properties, which provides a feasible approach for high-temperature composite applications.
Footnotes
Acknowledgments
The authors gratefully appreciate the financial support from the National Natural Science Foundation of China (grant numbers 52473075, 52173074) and the Fundamental Research Funds for the Central Universities (grant numbers JKD01261701).
Author contributions
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (grant numbers 52473075, 52173074) and the Fundamental Research Funds for the Central Universities (grant numbers JKD01261701)
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.