
Abstract
Phthalonitrile (PN) resin has attracted significant attention due to its exceptional thermal stability and mechanical properties. However, the requirement for elevated curing temperatures remains a critical challenge for their widespread practical applications. In this study, phthalonitrile containing branched cyanine (BPN) was blended with silicon-containing arylacetylene (PSA) to investigate their synergistic curing effects. The 10 wt% PSA formulation offers an optimal trade-off, demonstrating that moderate PSA loading synergistically enhances both processability and thermal stability without compromising high-temperature mechanical robustness: a reduction of the initial curing temperature from 234.3°C to 198.7°C and a decrease in the melting point from 161.6°C to 147.4°C. Conversely, the processing window expanded from 41.3°C to 54.7°C. Furthermore, the system exhibited enhanced thermal stability, evidenced by an increase in the 5% weight loss temperature (T d5 ) from 530.33°C to 552.38°C and a rise in the char yield from 75.73% to 80.13%. No distinct glass transition was detected below 400°C, indicating a substantial improvement in both the processability and thermal stability of the blend system. Although the incorporation of PSA resulted in a slight reduction in the mechanical properties of BPN, the blends retained excellent mechanical performance at elevated temperatures, surpassing the room-temperature properties of PSA. The findings significantly enhance the industrial viability of phthalonitrile-based materials, effectively paving the way for their large-scale deployment in high-performance sectors.
Keywords
Introduction
In recent years, the escalating demand for improved thermal performance in resin-based composite materials for the aerospace and electronic packaging industries has drawn significant attention to high-performance resins.1–6 Phthalonitrile (PN) resins, for instance, are widely utilized across various fields owing to their outstanding thermal stability and flame retardancy. However, challenges such as high initial curing temperatures, narrow processing windows, harsh processing conditions, and high energy consumption limit their potential for industrial applications.7–9
To mitigate the issue of high initial curing temperatures in PN resins, incorporating high-performance resins or inorganic fillers via blending or copolymerization has emerged as a common strategy. This approach is designed to lower the melting point and widen the processing window.10–12 Song et al. incorporated an aluminum oxide into the resin matrix, successfully reducing the melting point from 237°C to 212°C and the initial curing temperature from 265°C to 228°C. However, the initial curing temperature remained above 200°C, necessitating high processing temperatures. 13 Weng et al. employed a composite curing agent containing melamine and ZnCl2 to cure resorcinol-based phthalonitrile. An initial curing temperature of below 200°C and a peak curing temperature of 217°C were successfully achieved in the blends. However, it was observed that the decomposition of melamine was easily induced at high temperatures, leading to the formation of structural defects within the material. 14 Liu et al. mixed silicon-containing phthalonitrile (Si2PN) with boron-modified phthalonitrile (BPN) to prepare a blend resin. A lower initial curing temperature was imparted to the blend by the introduction of BPN, which was accompanied by an enhancement in thermal stability (with T d5 being elevated from 430.3°C to 525.1°C). However, the large-scale industrial application of this system was restricted by the complex synthesis process of the resins. 15 In summary, developing efficient and practical strategies to lower melting points and broaden processing windows of phthalonitrile resins constituted a major focus in research on high-temperature-resistant phthalonitrile systems.
In previous work by our group, Hu et al. synthesized a silicon- and alkynyl-containing phthalonitrile (SiBEPN) with both silicon and an internal alkyne structure in the main chain. This modification effectively reduced the system viscosity (from 24 Pa·s to 10 Pa·s at 165°C) and lowered the melting point (from 211°C to 165°C), while maintaining good thermal stability (T d5 of 530°C). However, an initial curing temperature above 200°C was still required. 16 Separately, Liu et al. prepared a phthalonitrile containing branched cyanine (BPN); although the cured resin exhibited excellent heat resistance (T d5 of 540°C) based on high-density cyano, its practical application was limited by a high melting point of 182.7°C and an initial curing temperature exceeding 200°C, resulting in a processing window insufficient for actual working conditions. 17
Silicon-containing arylacetylene resins (PSA) are a class of high-performance thermosetting resins developed by East China University of Science and Technology.18–20 The introduction of Si-C bonds and arylacetylene into the backbone imparts outstanding heat resistance to the resin system, realized through the cross-linking of terminal or internal alkyne groups. Notably, PSA resins bearing vinyl side groups exhibit a liquid state at room temperature thereby achieving excellent fluidity.
Building on these studies, we proposed a blend strategy incorporating PSA into the BPN system. The introduction of PSA is expected to disrupt the crystal lattice structure, thereby lowering the system viscosity. By leveraging the strong exotherm from terminal alkynes reactions near 160°C, the curing of nitrile groups is promoted at reduced temperatures. Consequently, the blend system achieves a lower initial curing temperature, which expands the processing window and significantly reduces the energy required for molding. Additionally, incorporating PSA is anticipated to further enhance the heat resistance of BPN.
The viscosity-temperature behavior and its effect on the processability of the blend system were evaluated through rheological measurements. Curing behavior was investigated using differential scanning calorimetry (DSC) and in situ Fourier transform infrared spectroscopy (FT-IR). Moreover, the improvement in thermal stability upon PSA incorporation was assessed by thermogravimetric analysis (TGA). Finally, the mechanical properties of quartz fiber-reinforced composites including flexural strength and interlaminar shear strength (ILSS) were also characterized.
Experimental section
Materials
N,N-dimethylformamide (DMF, analytical grade, ≥99%), tetrahydrofuran (THF, analytical grade, ≥99.8 %), and 4,4′-diaminodiphenyl sulfone (DDS) were all purchased from Shanghai Titan Technology. Quartz fiber (QF) used as the reinforcement was supplied by Hubei Feilihua Quartz Glass Co. Ltd (Jingzhou, China), with an areal density of 136 g·m−2 and a thickness of 0.14 mm. The phthalonitrile containing branched cyanine (BPN) and silicon-containing arylacetylene resin (PSA) were lab-synthesized at East China University of Science and Technology according to previous reports.17,21 The structures of PSA, BPN, and DDS were shown in the Figure 1. Structure of BPN, PSA and DDS.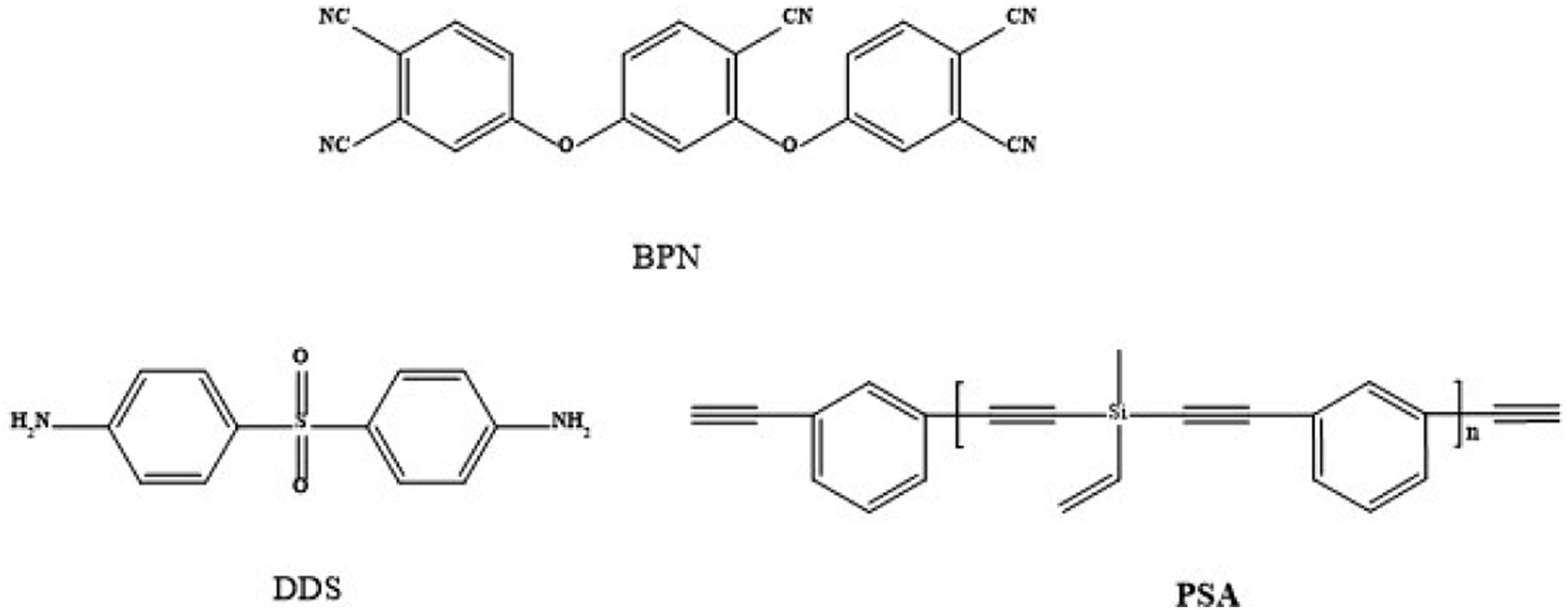
Characterization
Fourier transform infrared (FT-IR) spectra were acquired using a Nicolet 6700 spectrometer with KBr pellet preparation, scanned over 4000 ∼ 400 cm−1. For in-situ FT-IR, samples were held at 120°C, 170°C, 210°C, 230°C, 260°C and 300°C for 1 h each after heating at 5°C/min, with spectra collected every 15 min.
Rheological properties were measured with an RL-MARS3 rheometer (Thermo Hakke) from 25 to 240°C at a heating rate of 2°C/min under a shear rate of 0.01 s−1.
Differential scanning calorimetry (DSC) was performed on a Q2000 differential scanning calorimeter (TA Instruments) heated from 25°C to 350°C at 10°C/min in a nitrogen atmosphere.
Thermogravimetric analysis (TGA) was performed on a Mettler-Toledo TGA/DSC 1LF (Mettler-Toledo) instrument under nitrogen (flow rate: 60 mL/min), heated from 50 to 800°C at 10°C/min.
Dynamic mechanical analysis (DMA) was carried out using a DMA1 analyzer (Mettler-Toledo) from 50 to 350°C at 3°C/min and 1 Hz under nitrogen.
Flexural strength and interlaminar shear strength (ILSS) were tested at room and elevated temperatures using a CCSS-44100 electronic universal testing machine (Changchun Institute of Mechanical Science Co, Ltd), following standards of JC/T 773-2010 and GB/T 1449-2005, respectively; high-temperature tests included 10-min equilibration in a furnace.
Morphology was observed with an S-4800 field emission scanning electron microscope (Hitachi), with samples sputter-coated with gold for >60 s.
X-ray diffraction (XRD) analysis was carried out using a SmartLab 9 kW X-ray diffractometer (Rigaku) from 2θ = 3° to 50°.
X-ray photoelectron spectroscopy (XPS) was performed on a Thermo Scientific K-Alpha+ (Thermo Fisher) with a magnesium X-ray source.
Preparation of PSBPN resins
Compositions of specimens.
Preparation of BPN and PSBPN thermosets
Blends with varying weight ratios were cured in a muffle furnace according to the following schedule: 120°C/2 h, 170°C/2 h, 210°C/2 h, 230°C/2 h, 260°C/1 h, 300°C/1 h, 335°C/1 h and 375°C/6 h. After cooling naturally to room temperature within the furnace, black cured products were obtained.
Preparation of QF/BPN and QF/PSBPN composites
Quartz fiber fabrics were impregnated with the resin system, keeping the resin-to-quartz fiber mass ratio at 35:65. The impregnated fabrics were first air-dried in a fume hood to remove most of the solvent and then transferred to a vacuum oven at 60°C to eliminate residual solvent, yielding QF/BPN, QF/PSA and QF/PSBPN prepregs.
The prepreg layers were loaded into a platen vulcanizing press and heated until the resin was fully melted. Residual solvent was removed by repeated pressure release cycles. Stabilization of the pressure indicated resin flow had largely ceased. The pressure was then maintained at 1.5 MPa, and curing proceeded according to the following schedule: 170°C/2 h, 210°C/2 h, 230°C/2 h, 260°C/1 h, 300°C/1 h, 335°C/1 h and 375°C/6 h. After curing and natural cooling, QF/BPN, QF/PSA and QF/PSBPN composites were obtained.
Results and discussion
Rheological properties of BPN and PSBPN resins
The processability of the PSBPN blend systems with varying PSA contents was first evaluated by rheological measurements (Figure 2). A viscosity threshold of 50 Pa·s was adopted to determine the melting point and gelation temperatures. Compared to the melting point of 161.6°C for neat BPN resin (with 5 wt% DDS), the blend resins showed a continuous decrease in melting point with increasing PSA content. For instance, the melting points reduced to 147.4°C for PSBPN90 and further to 144.2°C for PSBPN80. This depression is mainly attributed to PSA disrupting the ordered arrangement of BPN, thereby hindering molecular packing.
13
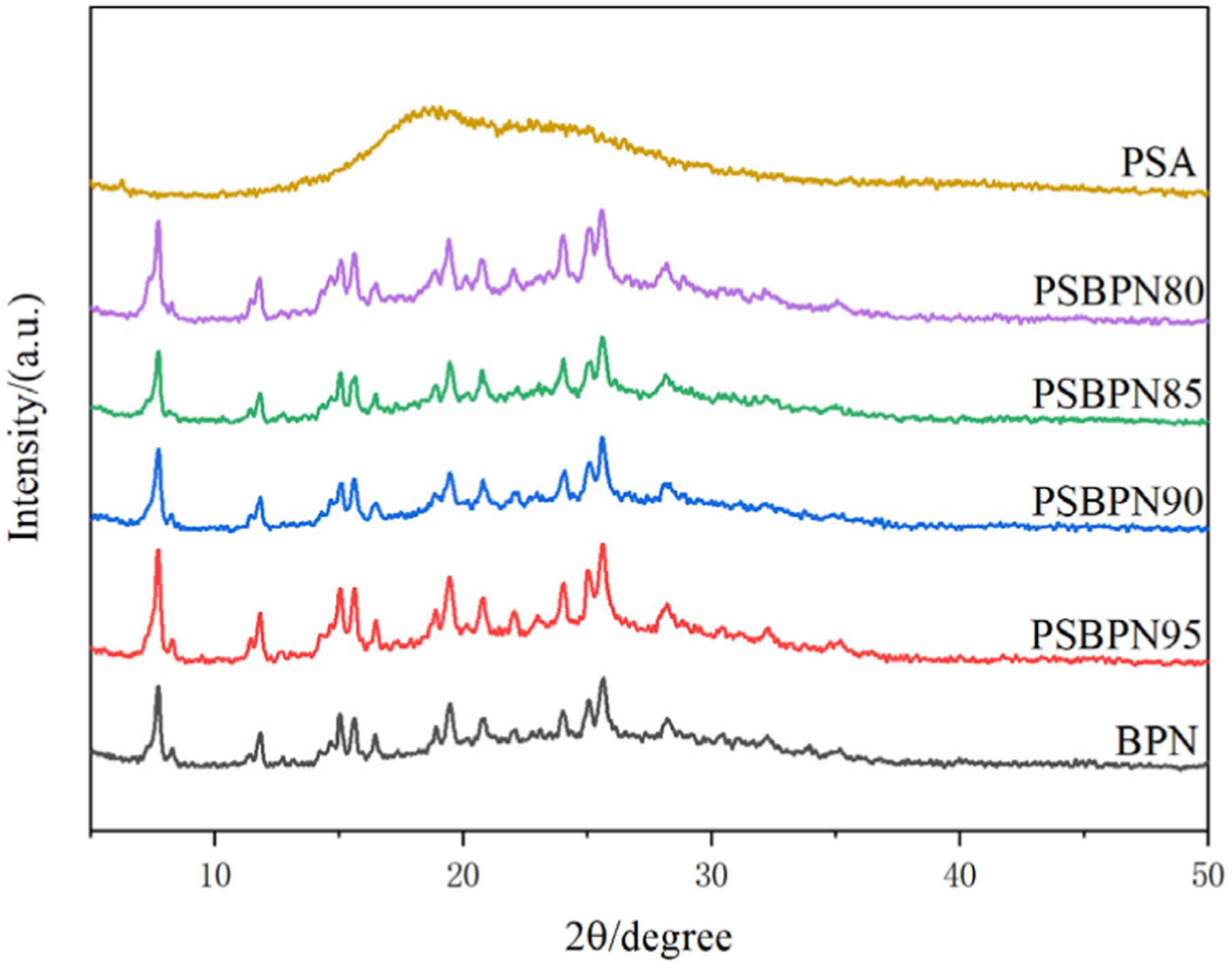
To explicitly verify this hypothesis, XRD analysis was performed on the blended resins with varying mass ratios (Figure 3). The relative crystallinity (X
c
) was quantitatively determined using the equation (1). Where A
c
and A
a
denote the integrated areas of the crystalline diffraction peaks and the amorphous halo, respectively. As shown in Figure 3, the neat BPN matrix exhibits a semi-crystalline nature with a relative crystallinity (X
c
) of 32.57%, whereas pure PSA is a typical amorphous liquid resin at room temperature (X
c
of 0%). With the gradual incorporation of PSA, although the characteristic diffraction peaks of BPN persist, the overall X
c
of the system decreases systematically: 29.70% for PSBPN95, 26.41% for PSBPN90, 25.06% for PSBPN85, and 24.45% for the PSBPN80 blend. This trend clearly indicates that the introduction of PSA effectively disrupts the highly regular lattice packing of pristine BPN, thereby suppressing the crystallization capability and lowering the melting point of the blended resin. Complex viscosities of BPN and PSBPN. XRD curves of BPN, PSBPN and PSA.
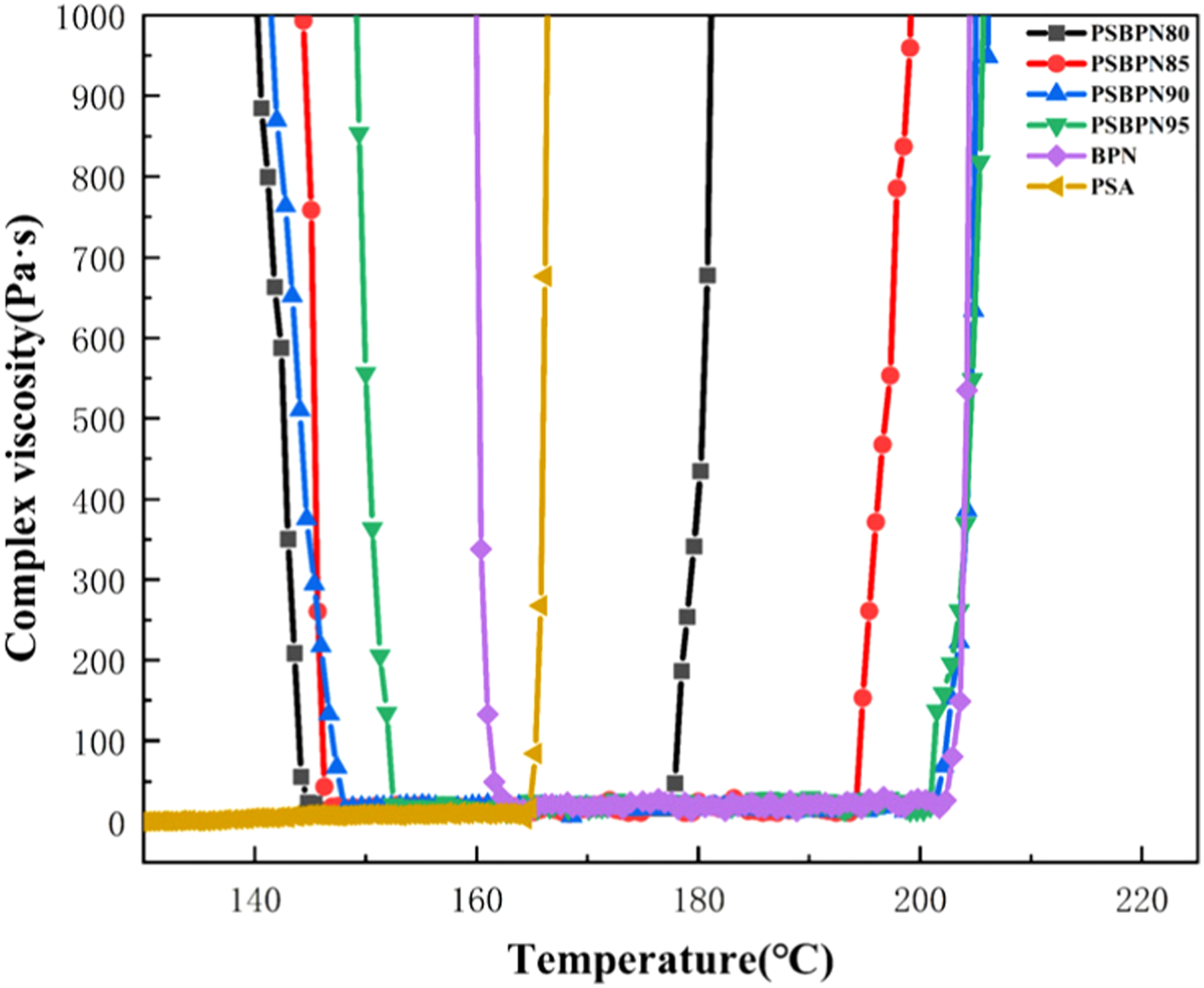
Similarly, relative to the gelation temperature of 202.9°C for neat BPN (with 5 wt% DDS), the blends exhibited a progressive drop in gelation temperature upon PSA addition, implying a corresponding decrease in the initial curing temperature. At 10 wt% PSA, the gelation temperature shifted to 202.1°C, and it decreased further to 178.5°C at 20 wt% PSA. This trend is ascribed to the high concentration of highly reactive terminal alkyne groups in PSA which undergo vigorous exothermic curing around 160°C, accelerating nitrile group cross-linking. The resulting early network formation causes system viscosity to rise rapidly at a lower overall temperature. 22
The processing window (the temperature range between melting and gelation) widened from 41.3°C for neat BPN to 48.4°C, 54.7°C, and 47.7°C at PSA contents of 5 wt%, 10 wt%, and 15 wt%, respectively, but narrowed to 34.3°C at 20 wt% PSA. The processing window shows a trend of rising initially and then falling. This non-monotonic change reflects competing factors. At lower PSA contents, melting point depression dominates and expands the window. At higher loadings, the pronounced drop in gelation temperature due to alkyne curing becomes dominant, narrowing the available range. Overall, the 10 wt% PSA formulation offers the best balance, combining a reduced melting point (147.4°C vs 161.6°C), a wider processing window (54.7°C vs 41.3°C), and a low system melt viscosity (∼25 Pa·s).
Curing mechanism of PSBPN resin
To clarify the curing behavior of the blend system, particularly the reduction in initial curing temperature, the thermal transitions during resin curing were analyzed by differential scanning calorimetry (DSC) as shown in Figure 4 and Table 2. Neat BPN with the addition of DDS showed a melting point of 167.4°C and a curing peak at 256.4°C. Its relatively moderate cured performance compared to literature values was attributed to the formation of a 4,4′-oxydiphthalonitrile during synthesis.
17
This structure unit is intentionally retained because it lowered the melting point and benefitted subsequent molding. (a) DSC curves of the resins (b) DSC peak-fitting of PSBPN. Rheology, DSC and TGA results of resins.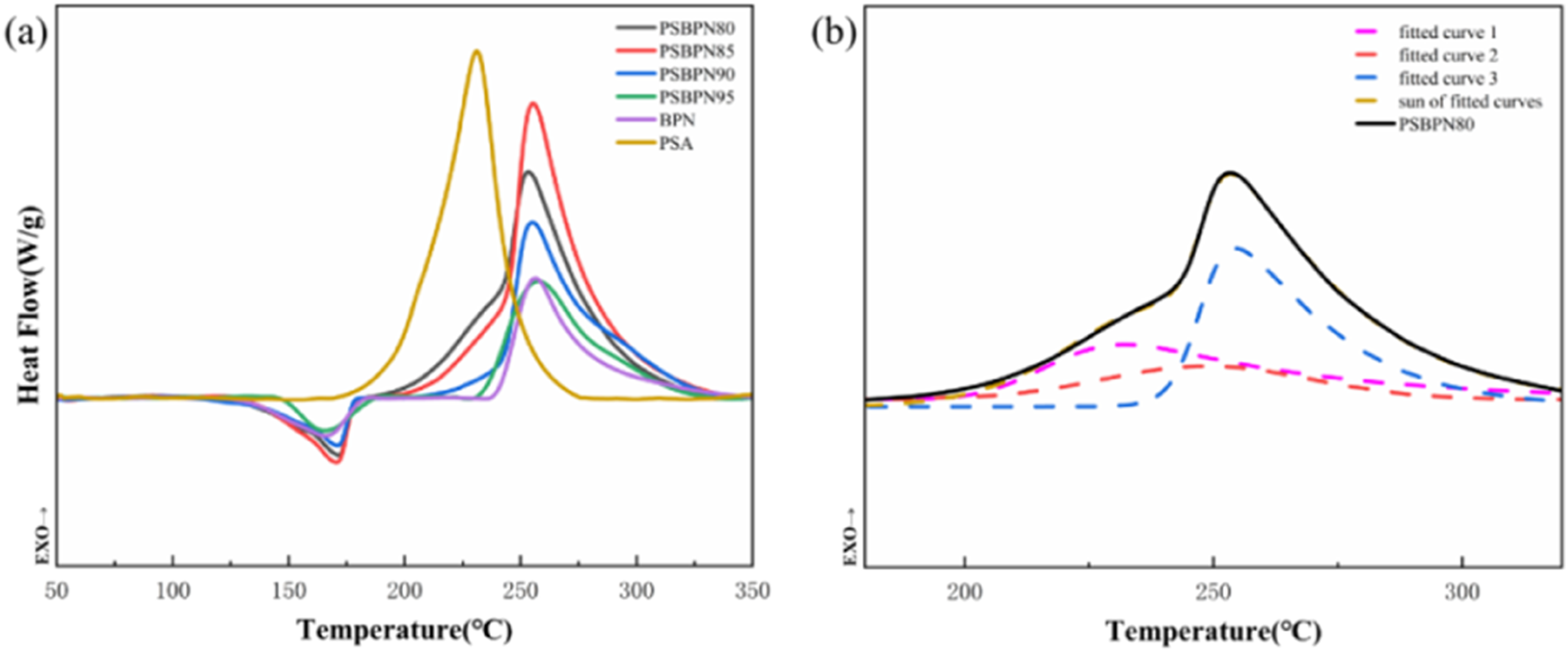
A slight mismatch between DSC melting points and rheological melting temperatures was noted and ascribed to overlapping thermal events. The exotherm from PSA curing near the melting region partially offset the endotherm from resin melting. Pure PSA exhibited an onset curing temperature of 168.7°C and a curing enthalpy of 626.1 J·g−1, whereas BPN/DDS had an onset of 234.3°C and an exothermic enthalpy of 203.5 J·g−1. As PSA content increased, the blend’s onset curing temperature decreased while the total exothermic heat rose. At 20 wt% PSA, the onset dropped from 234.3°C to 182.8°C and an exothermic enthalpy increase from 203.5 J·g−1 to 313.3 J·g−1, confirming that PSA promotes BPN curing, consistent with earlier rheological shifts to lower gelation temperatures. This acceleration is attributed to substantia heat release from alkyne curing, which initiates nitrile polymerization at lower external temperatures. 23
Peak deconvolution of PSBPN curves (Figure 4(b)) revealed that fitted peaks did not simply match those of individual components. The BPN curing peak shifted forward, further supporting PSA-enhanced initiation. Simultaneously, the PSA cross-linking peak broadened, likely due to restricted molecular mobility imposed by BPN’s initial network. 24 An additional exothermic peak suggests a synergistic reaction pathway involving both alkyne and cyano groups. 25
To assign the reaction responsible for the third exothermic peak, in situ FT-IR monitoring of PSBPN80 was conducted (Figure 5). Absorption bands at 3300 cm−1 and 2148 cm−1 were assigned to C–H stretching of terminal alkynes (external alkynes) and (–C≡C–H) and C≡C stretching of internal alkynes in PSA, respectively. Alkyne reactions in such systems are known to proceed mainly via cyclotrimerization to benzene rings, addition forming conjugated polyenes, and Diels–Alder reactions yielding biphenyl and naphthalene structures.26–29 In situ FTIR traces of PSBPN80 monomer heated from 25 to 300°C.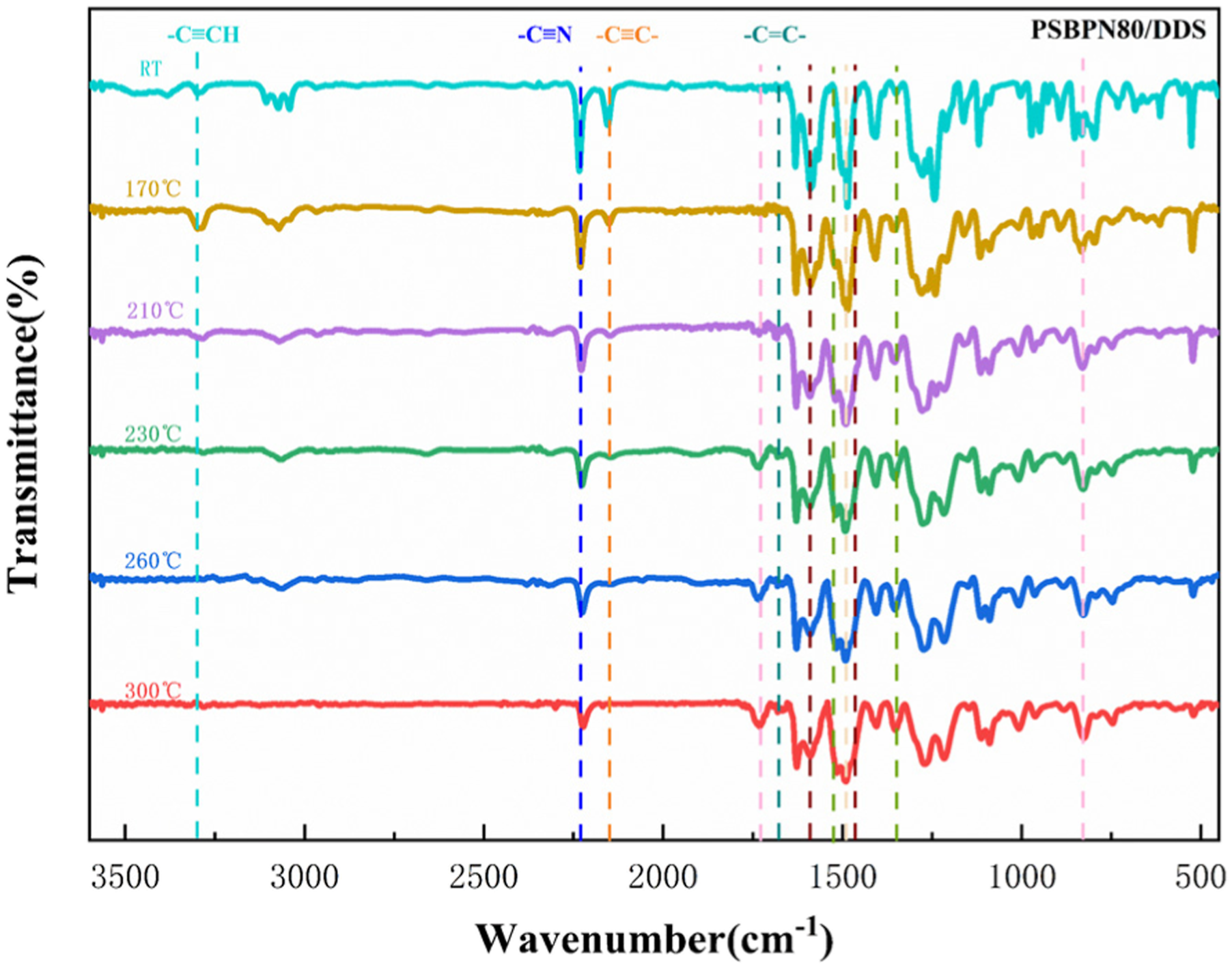
The band at 2233 cm−1 was assigned to the cyano group. Its intensity gradually diminished with rising temperature. Concurrently, emerging absorptions at 1723 cm−1 and 835 cm−1 were attributed to isoindoline structures, while signals at 1525 cm−1 and 1350 cm−1 corresponded to triazine rings, and features at 1016 cm−1 indicated phthalocyanine formation. All of these groups were recognized as key structures in phthalonitrile curing.30–32 Crucially, the increase in isoindoline-related peak intensities was more prominent than those for triazine and phthalocyanine rings below 300°C, suggesting that cyano conversion initially favors isoindoline formation prior to extensive heterocycle cross-linking. Together, these results supported a curing process in which PSA and BPN undergo largely independent self-curing, reactions, generating an interpenetrating cross-linked network.
Notably, a band at 1470 cm−1 emerged uniquely in the blend systems and was assigned to pyridine ring skeletal vibrations, confirming cyclotrimerization between alkyne and cyano groups to form pyridine heterocyclic structures. This observation directly validated the deconvolution-based inference from DSC regarding a distinct synergistic exotherm. 33
To further verify the formation of pyridine rings, X-ray photoelectron spectroscopy (XPS) was conducted on the cured PSBPN80 blend, and the high-resolution C 1s and N 1s spectra were systematically deconvoluted (Figure 6). Compared with the XPS spectrum of the cured pure BPN,
34
no distinct or independent peak corresponding to the newly formed pyridine rings is observed in the C 1s spectrum (Figure 6(a)), primarily due to severe overlap in binding energies. The peak centered at 285.70 eV is attributed to the combined contributions of residual unreacted cyano groups (-C≡N) and the carbon-nitrogen double bonds (C = N) within the pyridine rings. Meanwhile, the peak at 286.83 eV, typically assigned to Ar-O/C-N) species, is consistent with the characteristic carbon environments in the triazine and phthalocyanine rings. C 1s (a) and N 1s (b) spectra of cured PSBPN80.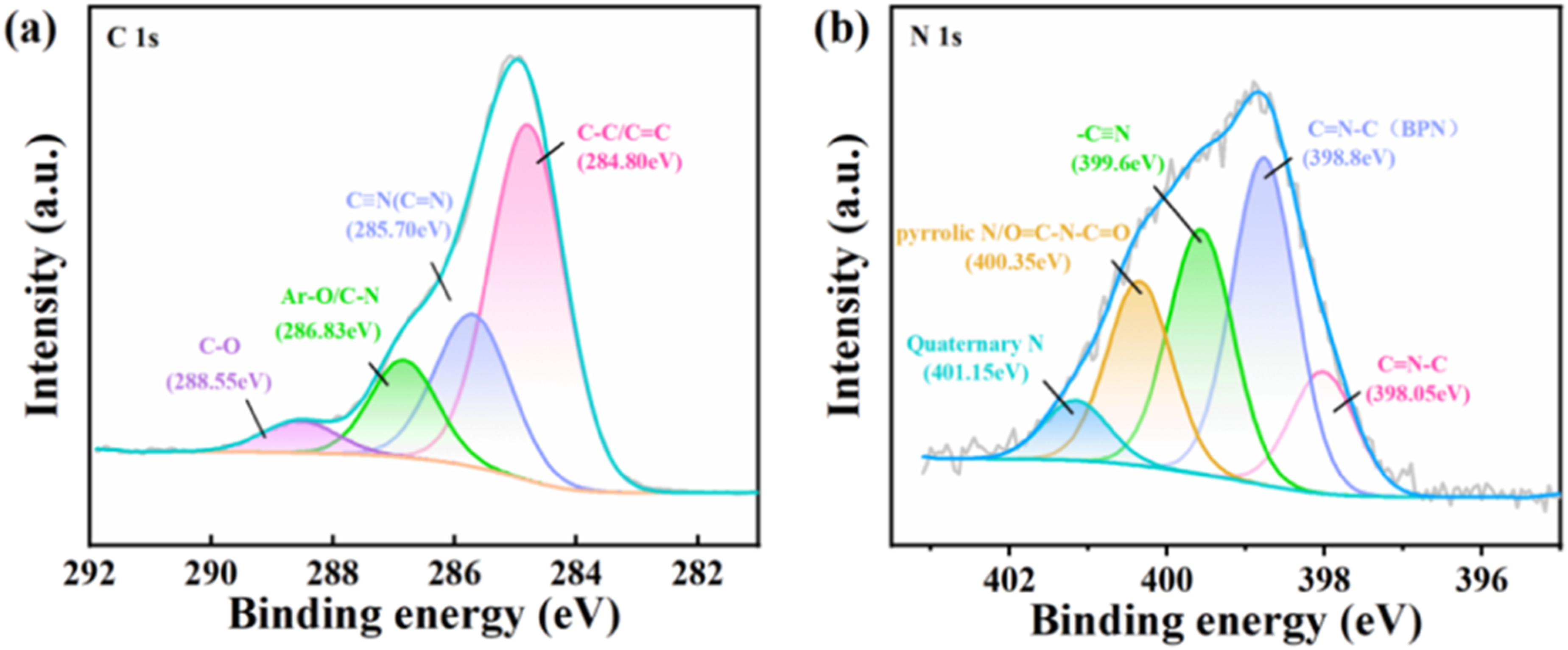
These assignments are further supported by the high-resolution N 1s spectrum (Figure 6(b)). In addition to the retained peak at 398.8 eV, which corresponds to C = N–C species arising from the polymerization products of BPN (isoindoline, triazine, and phthalocyanine rings), a distinct new peak emerged at 398.05 eV. Based on chemical shift principles, the triazine ring contains three highly electronegative nitrogen atoms, whose strong electron-withdrawing effect elevates the binding energy. In contrast, the pyridine ring contains only a single nitrogen atom, preserving a relatively higher local electron cloud density and thus exhibiting a lower binding energy. Accordingly, the new peak at 398.05 eV is unambiguously assigned to pyridinic nitrogen (Pyridinic N). These XPS findings are in excellent agreement with the in-situ FTIR results, providing strong evidence for the formation of pyridine rings via the cyclotrimerization between the alkynyl groups of PSA and the cyano groups of BPN.
Based on the combined analyses of DSC, rheological, and in situ FT-IR, the curing mechanism of PSBPN is proposed to center on concurrent cross-linking of C≡C and C≡N groups, as depicted in Figure 7. Alkyne groups primarily undergo three main types of reactions: cyclotrimerization (yielding benzene rings), addition reactions, and Diels–Alder reactions (yielding polyene and naphthalene frameworks). Cyano curing, initiated by active hydrogen from DDS amine groups, preferentially forms isoindoline structures around 300°C. At higher temperature, the terminal active hydrogen from isoindoline propagates further cyano cross-linking, generating heat-resistant heterocycles like triazine and phthalocyanine rings and substantially increasing crosslink density. Simultaneously, cyclization reaction between alkyne and cyano groups produces pyridine heterocycles, corroborating the distinct synergistic exotherm observed in DSC. Curing mechanism of PSBPN.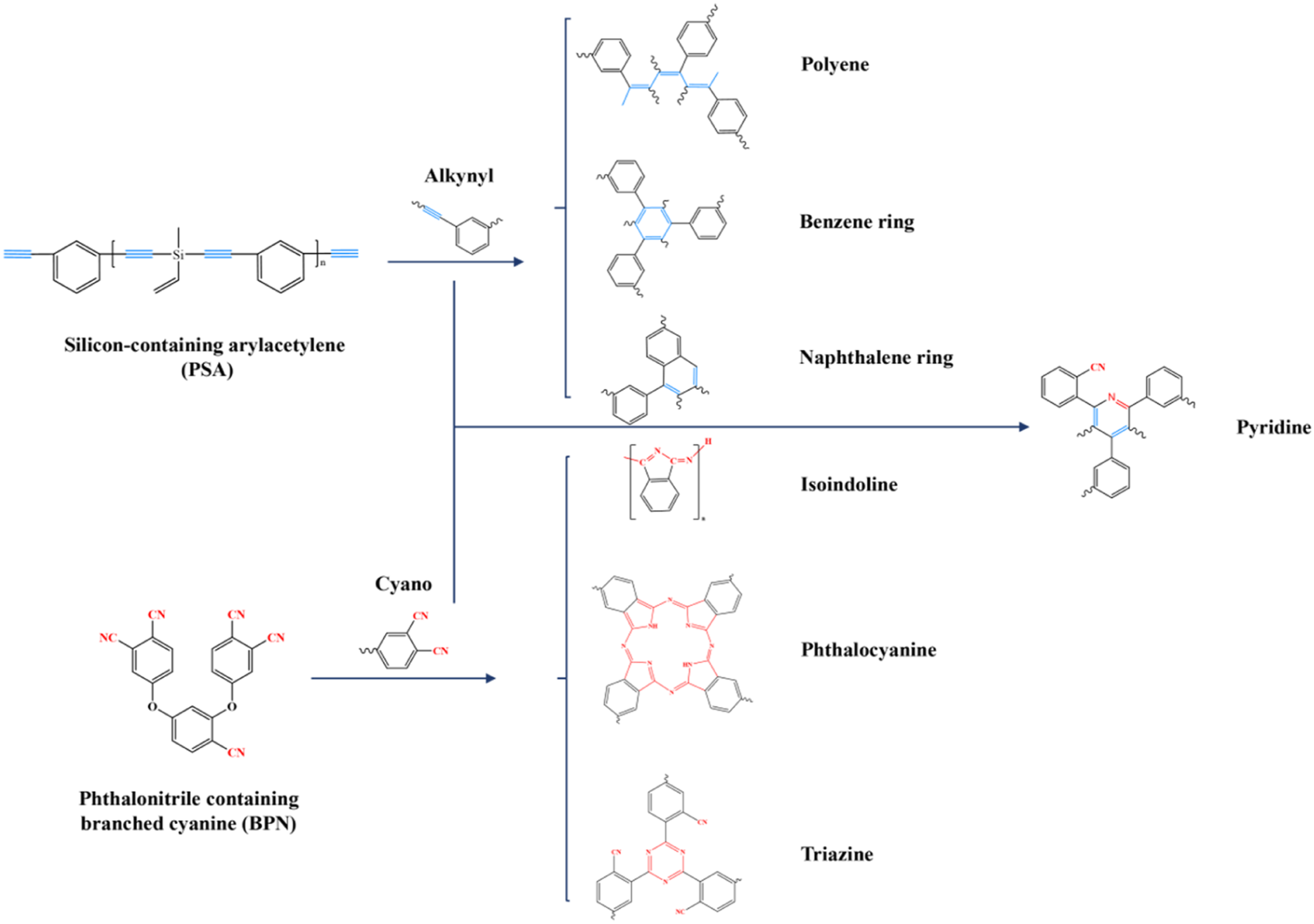
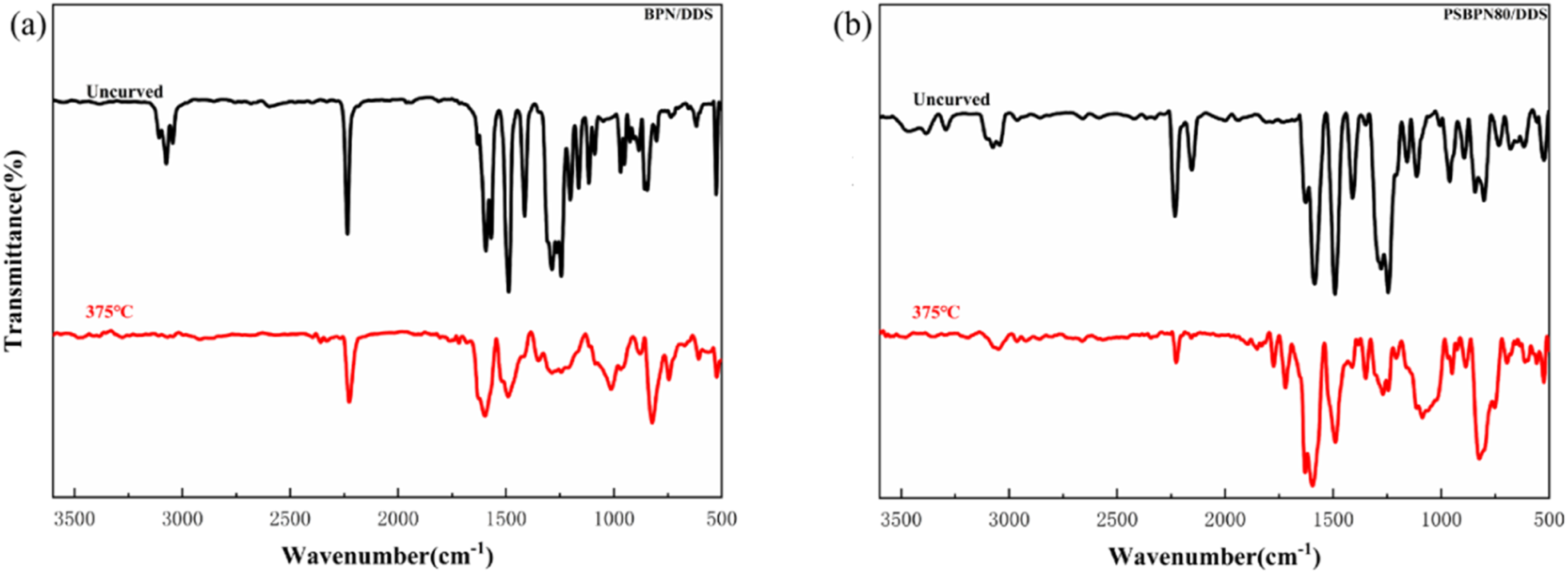
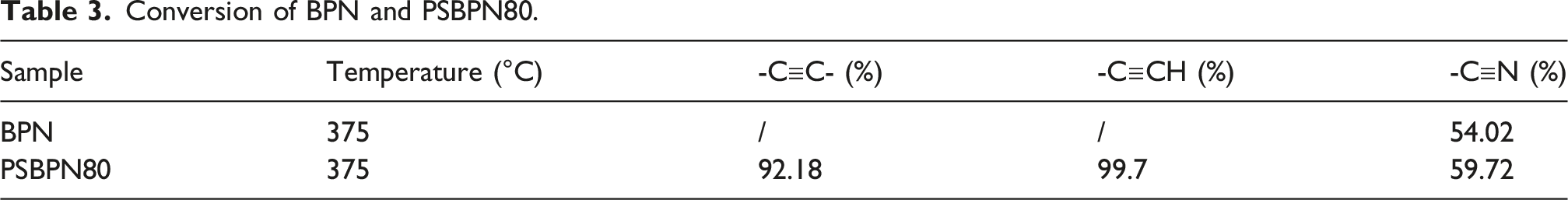
Despite instrument limitations, residual cyano groups were still detectable at 300°C in the in situ FT-IR traces. Additionally, prior work indicates that the main exotherm linked to phthalocyanine formation occurs above 350°C. To quantify group conversion, changes in characteristic FT-IR bands before and after full curing (375°C) were compared for representative BPN and PSBPN80 systems presented in Figure 8. For PSBPN80, the Si–CH3 stretch at 1243 cm−1 which unchanged during curing served as an internal standard, and conversions of the corresponding reactive groups were calculated via equation (2). For BPN, the benzene ring skeleton vibration at 1490 cm−1 was used as the reference to normalize cyano conversion. Here, A
i
/A
r
denotes the fractional conversion, and A
i
/A
r
is the peak area ratio of the target reactive group relative to the reference group at a given temperature. FTIR spectra of uncured resins and resins cured at 375°C. (a) BPN, (b) PSBPN80.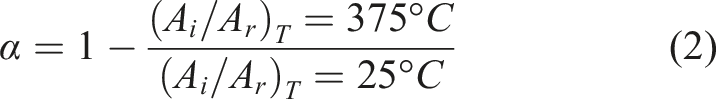
Conversion of BPN and PSBPN80.
Thermal properties of BPN and PSBPN resins
Thermogravimetric curves of cured BPN, PSA, and PSBPN cured resins at different blend ratios in a nitrogen atmosphere are shown in Figure 9. A minor weight loss below 200°C was observed, ascribed to residual cyano groups in BPN. As indicated by FT-IR, the cyano groups were not fully cross-linked. During grinding process, they readily formed hydrogen bonds atmospheric moisture. Subsequent desorption of this absorbed water during TGA heating produced the rapid initial weight loss.
17
TG curves of cured resins.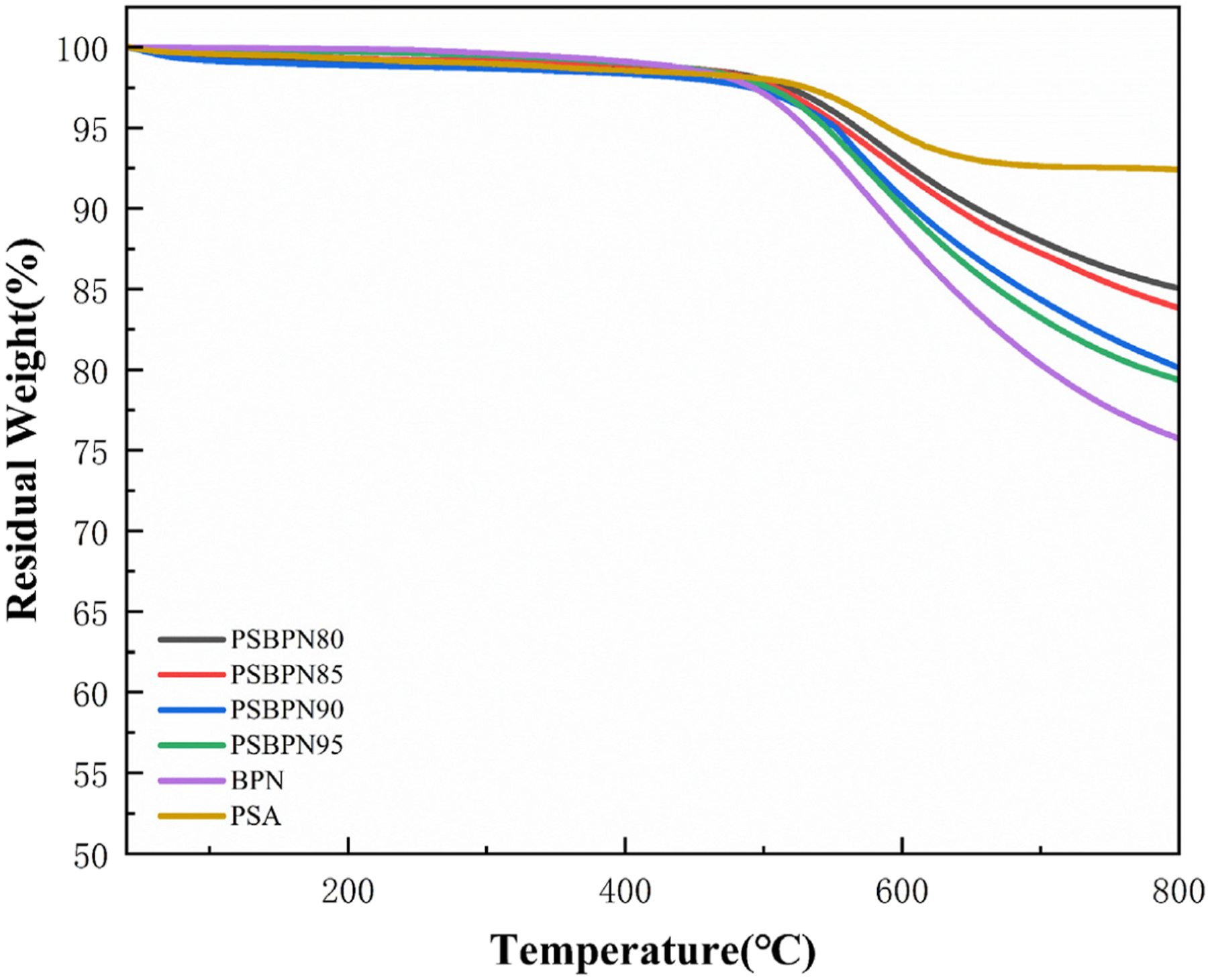
Temperatures at 5% weight loss (T d5 ) and residual weight at 800°C (Y r800 ) under nitrogen were compiled in Table 2. Neat BPN gave T d5 and Y r800 values 530.33°C and 75.73%, respectively, while PSA showed higher stability with T d5 = 588.15°C and Y r800 = 92.4%. Introducing PSA improved the thermal stability of PSBPN blend resins relative to BPN. At 20 wt% PSA (PSBPN80), T d5 rose to 567.55°C, and Y r800 increased to 85.15%. This enhancement stems from two contributions: (1) PSA promotes a higher curing extent of BPN by (consistent with DSC and FT-IR) yielding a denser crosslinked network, supplemented by the high-temperature alkyne-derived network from PSA itself. And (2) the silicon atoms in PSA chains decomposes at elevated temperatures to generate a protective C-Si-C ceramic layer, retarding further decomposition. 35
The glass transition temperature (T
g
) of QF/PSBPN composites cured at 375°C was evaluated by DMA and the results was shown in Figure 10. The composites retained a high modulus over a broad temperature range. However, the initial storage modulus decrease with increasing PSA content, attributable to the greater bond rotational freedom of Si-C bonds in PSA compared to C-C bonds, which increases free volume and thus lowers the storage modulus. Notably, the loss tangent (tanδ) of QF/PSBPN composite exhibits no distinct relaxation transition peak below 400°C, indicating a T
g
beyond the measurement range and confirming excellent high-temperature stability. DMA curves of QF fiber-reinforced composites.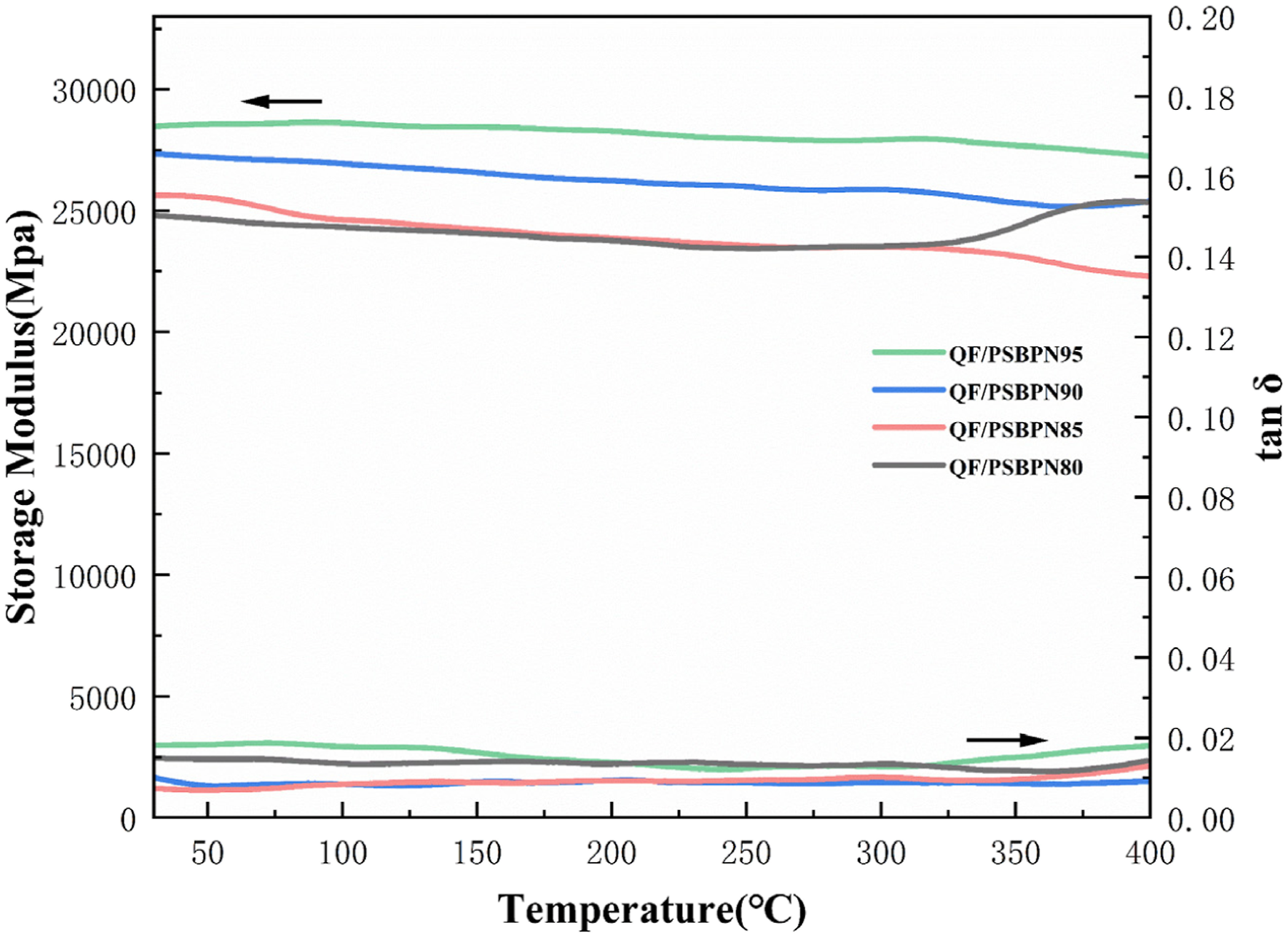
Mechanical properties of BPN and PSBPN composites
The flexural strength and interlaminar shear strength (ILSS) of BPN, PSA, and PSBPN composites with different blend ratios were measured at multiple temperatures, with the results presented in Figure 11. Neat PSA composites showed relatively low mechanical properties, with a flexural strength of 140.31 MPa and an ILSS of 12.27 MPa. In contrast, neat BPN resin composites exhibited superior mechanical performance with a flexural strength of 461.9 MPa and an ILSS of 38.55 MPa. Introducing PSA led to a gradual decrease in both properties for PSBPN blends. At 20 wt% PSA, the values declined to 357.31 MPa and 27.73 MPa, respectively. This reduction was attributed to PSA’s lower molecular polarity and surface energy, weakening interfacial bonding between PSA resin and reinforcement fibers and thereby impairing stress transfer from resin matrix to fibers.
36
Despite a minor reduction in mechanical strength, the blend composites retain robust mechanical properties at low PSA loadings, offering a viable strategy to synergistically enhance processability and thermal resistance. Mechanical properties of QF reinforced composites.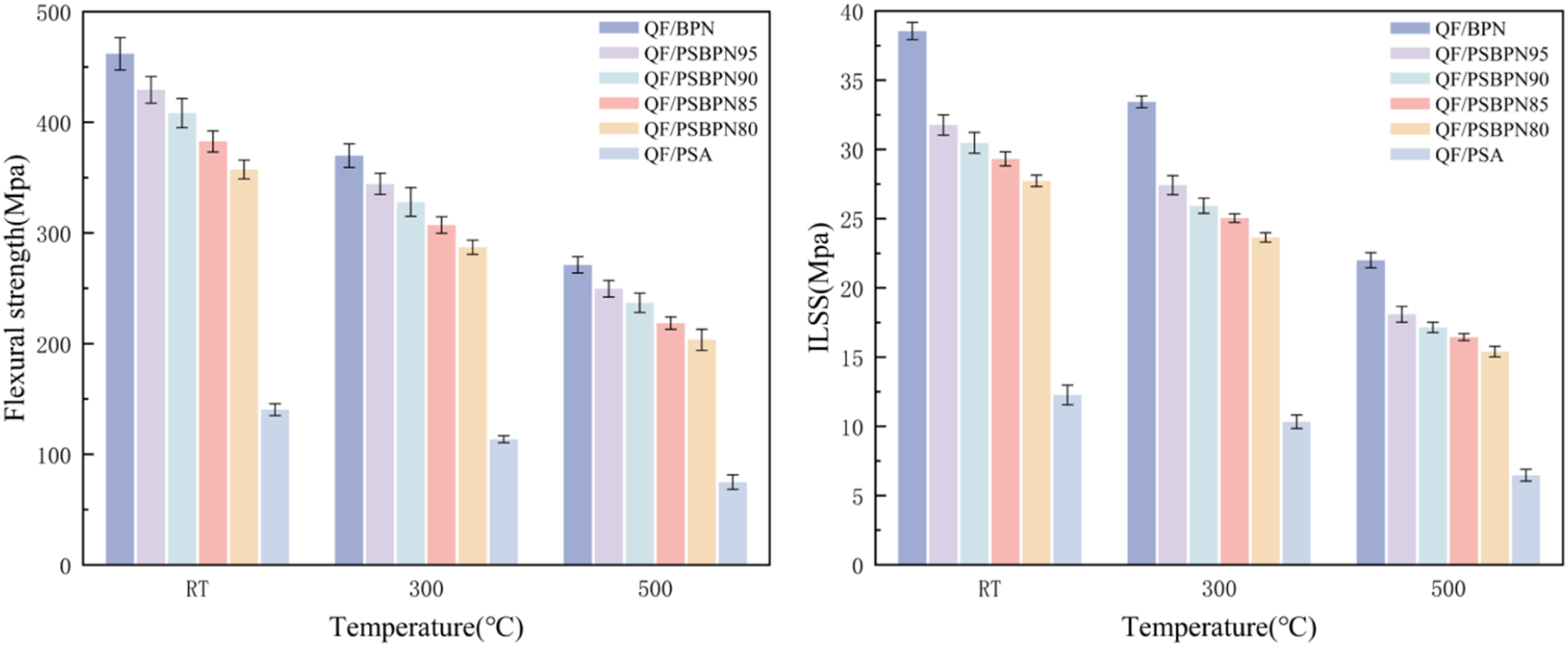
Based on a holistic evaluation of processability, thermal stability, and mechanical performance, 10 wt % was identified as the optimum composition. In the PSBPN90 system, the melting point decreased from 161.6°C to 147.4°C, while the onset curing temperature dropped from 234.3°C to 198.7°C, extending the processing window from 41.3°C to 54.7°C. Thermal stability improved concurrently: T d5 rose from 530.33°C to 552.38°C, and char residue at 800°C (Y r800 ) increased from 75.73% to 80.13%. No clear glass transition was detected below 400°C. Mechanical robustness was remained high, with flexural strength of 408.4 MPa and ILSS of 30.48 MPa, retaining exceeding 57.99% and 56.26% of these values at 500°C, respectively.
SEM analysis of composites fracture surfaces
To elucidate the mechanism behind mechanical properties degradation at elevated temperatures, the fracture morphologies of composites was examined by scanning electron microscopy (SEM). Figure 12 displays the fracture surfaces of resin tested at 25°C, 300°C, 500°C. It can be clearly observed that the composites exhibit excellent structural integrity at room temperature. Abundant resin adheres to the surface of the quartz fiber continuously, fills inter-fiber gaps, combined closely with fewer internal defects, which ensures the efficient stress transfer and excellent mechanical performance. After exposure to 300°C, some resin cracking and detachment occur, yet fiber–matrix adhesion remains sufficient to retain reasonable mechanical properties. At 500°C, however, the resin matrix undergoed marked thermal oxidation degradation and crosslinking network collapse. Fibers become largely exposed, and the matrix filling drops drastically, and widespread microcracks and voids form, accounting for the sharp decline in mechanical properties. SEM images of fractured surfaces of QF/PSBPN90 at different temperature: (a) 25°C; (b) 300°C; (c) 500°C.
Conclusions
This study introduced silicon-containing arylacetylene resin (PSA) into a phthalonitrile containing branched cyanine (BPN) resin to lower the initial curing temperature and improve its processability. Rheological analysis shows adding 10 wt % PSA reduced the melting point from 161.6°C to 147.4°C and widens the processing window from 41.3°C to 54.7°C. Excessive PSA 20 wt % narrows it to 34.3°C. DSC and FT-IR confirm PSA promotes cyano curing, lowering the onset curing from 234.3°C to 182.8°C. TGA and DMA reveal enhanced heat resistance of BPN with the addition of PSA: T d5 reaches 567.55°C, char yield at 800°C (Y r800 ) rose to 85.15%, and no distinct glass transition occurs below 400°C. In situ FT-IR identifies three curing pathways: alkyne cross-linking via Diels-Alder and addition reactions; cyano-derived heterocyclic structures (isoindoline, triazine, and phthalocyanine rings) catalyzed by DDS; and alkyne–cyano synergistic cyclization forming pyridine rings. Collectively, introducing 10 wt% PSA achieves a balanced system: melting and curing temperatures are significantly lowered, the processing window widens by ∼ 13°C, and thermal stability improves. The synergy originates from PSA disrupting BPN crystallinity, exothermic alkyne curing promoting earlier nitrile cross-linking, and alkyne–cyano cyclization generating additional pyridine networks. A modest PSA loading thus yielded a PSBPN system with significantly improved processability and high-temperature stability while retaining competitive mechanical properties. Though some mechanical strength was compromised, this route offered a practical strategy to engineer phthalonitrile resins balancing ease of processing with robust thermal performance.
Footnotes
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Yichen Guan, Hongfang Tang, Xiwei Liu, Jianke Hu, Zijian Ping, Yanhong Hu supported by the Fundamental Research Funds for the Central Universities (No. JKD01261701).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
The date that supports the findings of this study are available on request from the corresponding author upon reasonable request.