
Abstract
In the third issue of the series on modern quantum chemical methods in the support role of NIR spectroscopy we continue to introduce the researchers from the field of experimental spectroscopy to practical aspects and applications of modern anharmonic theoretical approaches. The first two issues focused on explaining the necessary theoretical and practical background, allowing readers to get more familiar with the topic. An overview of recent literature reports highlighted the advantages stemming from using quantum chemical calculation in the support role to NIR spectroscopy. These deliberations were based on several cases of small- to medium-sized molecules. This part overviews the topic of applications of quantum theoretical methods to complex molecules with practical significance, which typically prove to be challenging objects for theoretical studies. An exemplary application of presented methodology to the case of Rosmarini folium biological samples is also examined here. The rosemary specific active compound, rosmarinic acid, is a relatively complex polyphenol with growing phytopharmaceutical importance, and therefore provides an excellent object of applied studies. The possibilities of combining the information stemming from quantum chemical calculation with the methods of advanced spectral data analysis, which are commonly used in experimental NIR spectroscopy (chemometrics, two-dimensional (2D) correlation spectra) are also overviewed. Again, these deliberations are based directly on the most recent reports published in the field.
Introduction
This is the third part of the series of publications reviewing the quantum chemical methods in the role of a support tool for NIR spectroscopy. In the first two parts a general introduction to this topic was provided, and examples of applications to some basic molecules were discussed. In this part, we will overview the possibilities of application of anharmonic quantum chemical methods to complex molecules, which obviously are of special importance to NIR spectroscopy.2–3 We will briefly cover the main concerns regarding the treatment of relatively large molecular systems, explaining why these present particular difficulty in theoretical approaches. It is worth highlighting, that the topic of anharmonic calculations of complex molecules is very rarely discussed even in the most recent literature. 4 Moreover, experimental studies of complex molecules by NIR spectroscopy usually largely benefit from advanced data analysis methods, such as chemometrics or two-dimensional correlation spectroscopy (2D-COS). 5 In this paper, we will discuss completely new possibilities, stemming from combined approaches, involving both quantum chemical results and advanced data analysis, for obtaining deeper insight and thorough spectral information on complex samples. These aspects will be directly commented on the basis of the theoretical and experimental data obtained for rosmarinic acid (RA), 6 which is a moderately complex polyphenolic natural compound with growing importance in phytopharmaceutical industry, due to its anti-oxidant activities. In recent years, the importance of phytopharmacology vastly expanded, as active compounds obtained from natural resources offer significant advantages on multiple planes. This tendency in aiming for a better exploration of phytopharmaceuticals (‘green pharma-chemistry’) also complements well into a general trend of focusing on natural products, which presently can be widely witnessed. However, the use of natural products also introduces substantial difficulties, particularly because of variability in the composition of a natural resource. Above reasons rise a demand for an adequate analytical control of the product, without an excessive escalation of the analysis cost and finally involve particular challenges for the quality control/quality assurance (QC/QA). These concerns are focused on by Huck Lab, which develops state-of-the-art analytical approaches to phytopharmaceutical samples and explores applied NIR spectroscopy in an exceptional role of a non-invasive, fast and cost-efficient analytical method,7–12 excellent for both analytical lab and industrial use. In the present issue, we will also overview the recently emerging possibility of including substantial support from quantum theoretical methods into applied analytical studies of natural pharmaceutical samples.
Complexity of experimental NIR spectra of large molecules
As it is well-known, NIR spectra of any kind of organic molecules usually exhibit a substantial level of complexity. NIR data frequently proves to be notably difficult for detailed analysis, and it is the major reason for the application of advanced data analysis methods. Some common properties of NIR spectra, such as high level of band overlap, substantial number of contributing vibrations – both factors are significantly bigger than for IR region – remain among the main reasons for wide application of chemometrics in NIR spectroscopy.
The complexity of NIR spectra stems directly from a high number of bands arising from combination modes. While the number f of normal (fundamental) modes for any non-linear molecule (here we disregard the case of linear molecules, as these are obviously a non-issue in the deliberated topic of complex molecules) can be described by equation (1)


As can be seen the number of normal modes (vibrational levels of freedom) increases linearly with the number of atoms in the molecule. Obviously, the number of overtones of each order is the same as the number of normal modes. However, the number of binary combinations increases non-linearly, at square dependence of the number of atoms in the molecule. This leads to a steep increase in the number of binary combinations, which may be observed as experimental bands in the NIR spectra. Obviously, there are also higher order overtones and combination bands. Nevertheless, due to population of vibrational levels and the intensities of the corresponding bands, usually the first order (binary) combinations and first overtones have the major influence on experimental NIR spectra.
Note that due to selection rules not all of these bands may appear in the experimental spectra, however these deliberations should give a general insight on the discussed matter. In Figure 1, we present the dependence of the number of modes (fundamental and binary combinations) on the number of atoms in a non-linear molecule. This simple diagram should give an overall impression of how complex the problem of the treatment of theoretical NIR spectra can become for bigger molecular systems. For molecules up to around 20 atoms, the number of binary combination modes is about 25–30 times the number of fundamental modes, and already extends over 1400. However, with increasing number of atoms, we observe a tremendous increase in the number of binary combinations, reaching ten thousands, still for a relatively not so complex molecule containing around 50 atoms.
The dependence of the number of modes (fundamental and binary combinations) on the number of atoms in a non-linear molecule.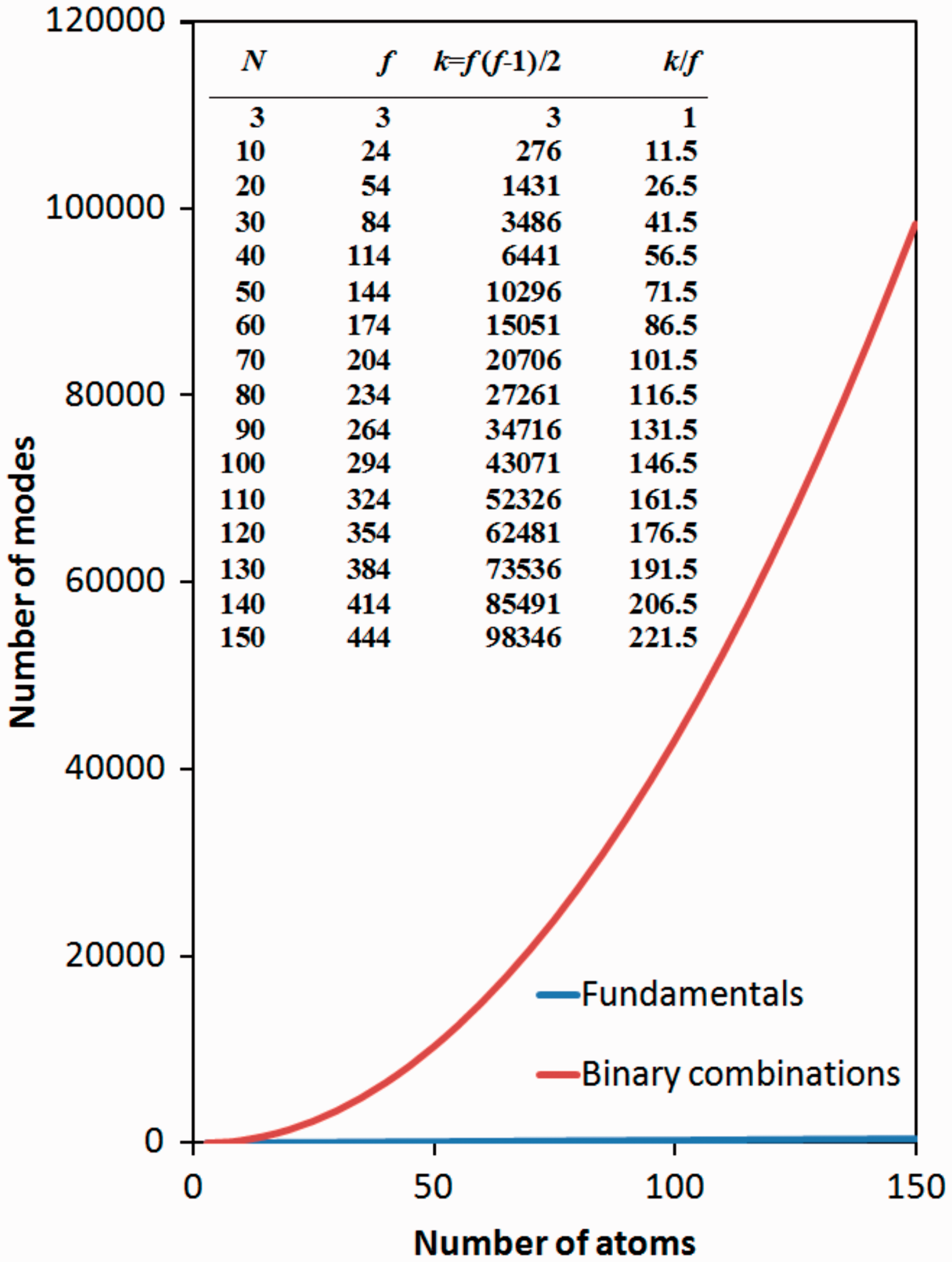
Such significant amount of contributing bands, which obviously tend to overlap heavily, often decreases our ability to conclude clearly from spectral line shape. For example it could be expected, that different modes may react differently to various external factors applied to the sample. Obviously, these circumstances explain the inherent complexity of NIR spectra, the difficulties in the analysis of NIR data and to some extent, inability to conclude strong spectra-structure correlations, at least when confronted with IR spectroscopy.
For the reasons explained above, even processing the calculated data proves not to be straightforward. In our procedure, we model each single calculated band by applying a bandshape function (usually a four-parameter Cauchy-Gauss product function). Afterwards, all modelled bands are summed over to form the final theoretical spectral envelope, which we compare with experimental spectrum. These procedures involve large datasets, which need to be successfully handled. A software suite reliable in processing relatively large datasets is recommended. In our studies, we prefer to use MATLAB package 13 for these tasks.
Application of anharmonic methods for the molecule of RA
For better depiction of the discussed matter, we present the data we obtained for RA6. RA is a polyphenolic compound that can be extracted from Rosmarinus officinalis L. and other plant samples, for example from Lamiaceae family. It has a remarkable antioxidant potential, due to existence of hydroxyl moieties which act as radical scavengers. Therefore, we recently can witness an increasingly growing importance of RA for phytopharmaceutical industry.
In a recent study, Kirchler et al. 6 studied Rosmarini folium samples; this work focused on multiple scientific problems, including an evaluation of analytical performance of different spectrometers as well as modern portable devices. However, the particular aspect that should be highlighted in the present review is the application of GVPT2 calculation for theoretical reproduction of NIR spectrum of RA. The results of theoretical study were subsequently used as an aid in the analysis of multivariate data analysis plots and 2D-COS synchronous plots. It is one the first reports on successful use of calculated NIR spectra in an applied study of a complex biosignificant sample.
The molecule of RA contains 42 atoms (Figure 2), which corresponds to 120 fundamental vibrations, 120 first overtones and 7140 binary combinations that were obtained through quantum chemical calculation. Therefore, compared to examples discussed in the two preceeding issues, RA molecule presents a more challenging object for an anharmonic quantum mechanical study. This should have a considerable impact on the complexity of the experimental NIR spectra of RA as well. Having previously (in the part two of the series) explained the correspondence of extensive resource demands of fully anharmonic calculation, it comes as no surprise that Kirchler et al.
6
have chosen efficient GVPT2 anharmonic approach on DFT level in their quantum chemical study of RA. The chosen density functional, single-hybrid B3LYP functional, offered good balance between computational cots and accuracy in that case. While double-hybrid functionals, such as B2PLYP, can often provide even better results, their application comes at a significant time resource cost, as recently evidenced by Beć et al.
14
In case of RA molecule, the use of double-hybrids would be extremely inefficient. Another important consideration, for the accuracy and efficiency of the calculation, is related to the choice of a basis set. The N07D basis set offers significant advantages in regard to both, as shown by Beć et al.15,16 As the reference experimental NIR spectrum of RA was measured for amorphous solid state sample, Kirchler et al.
6
obviously did not applied any solvent model in their study.
The molecule of rosmarinic acid. Molecular geometry optimized on DFT-B3LYP/N07D level of theory.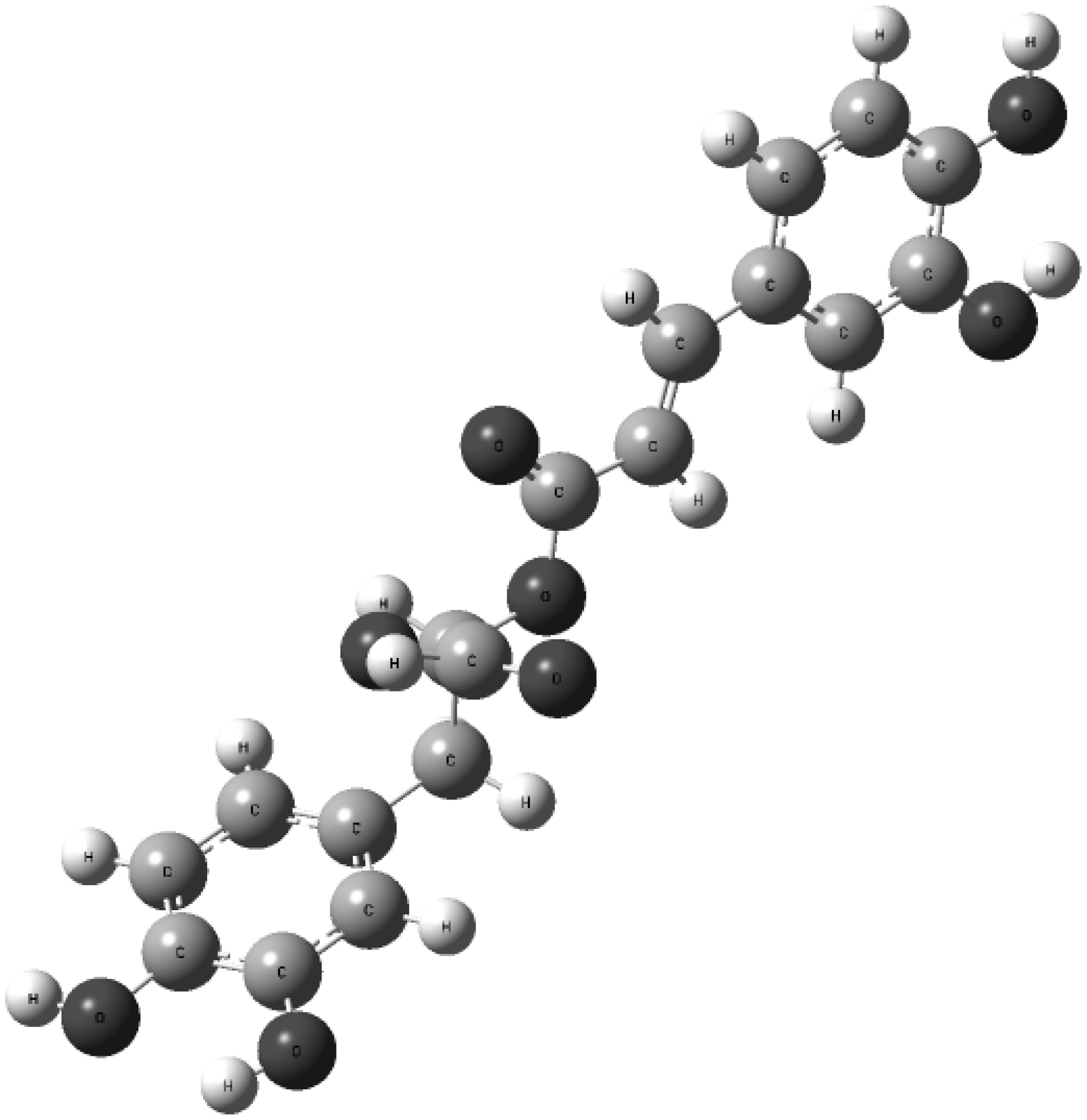
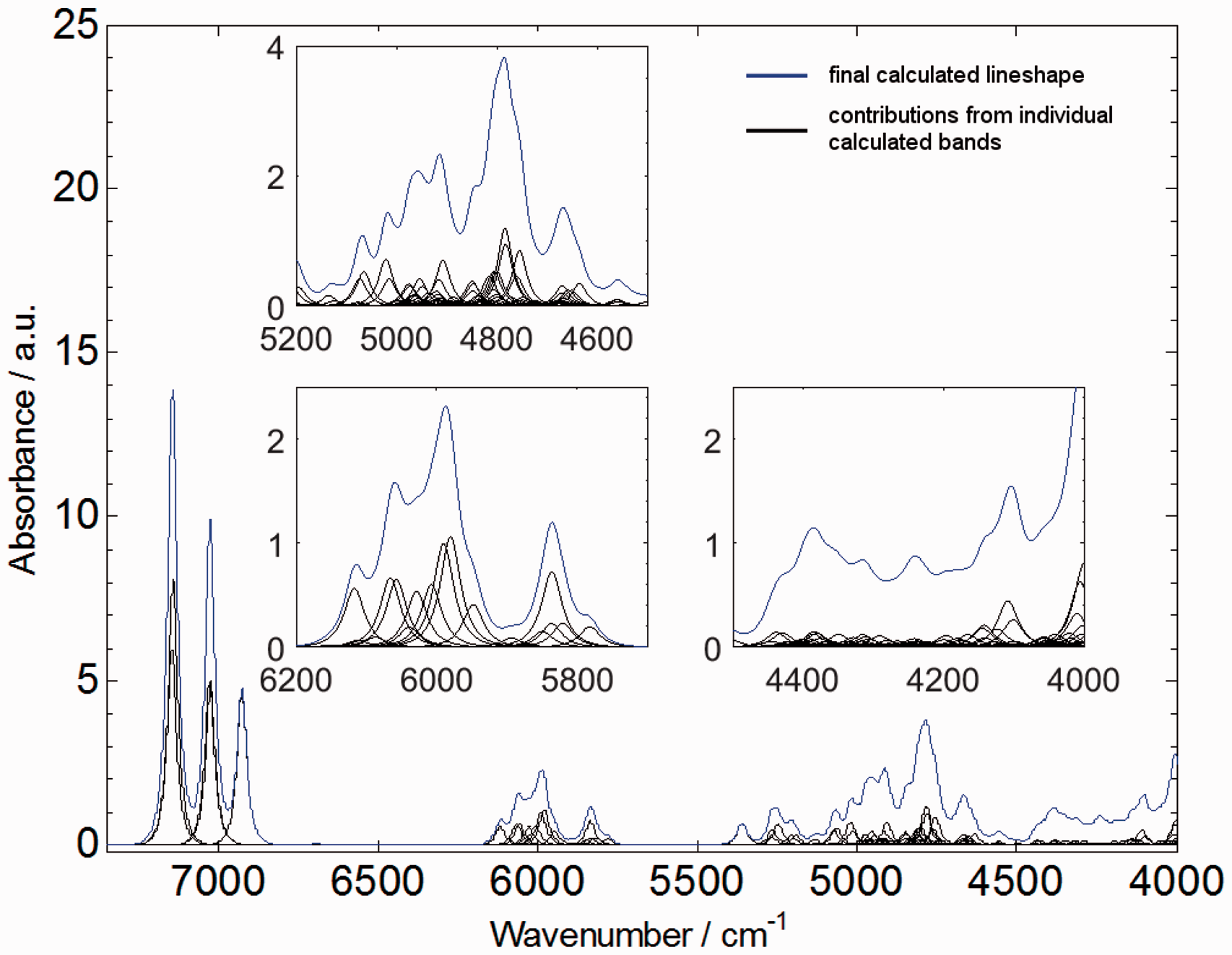
The results of the GVPT2-B3LYP/N07D calculations proved to be able to reproduce NIR spectrum of RA with a remarkable accuracy, taking into account the complexity of the RA molecule. The resulting theoretical NIR spectrum provides a detailed insight into formation of the experimental NIR spectrum. As presented in Figure 3 the overtone modes mainly contribute to the upper half (7250 to 5800 cm−1) of the RA spectrum, while the lower wavenumber region (below 5400 cm−1) is populated almost entirely by combination modes. Again, note that the standard GVPT2 approach provides the data on first overtones and binary combinations; however most often these modes play the major role in forming NIR spectra, due to higher intensities when compared to higher level overtones and combination modes. Looking further, the theoretical data provide remarkable evidence on the origin of the inherent complexity of NIR spectra, as presented in Figure 4.
The overall contribution of first overtones (green line) and binary combinations (black line) in the theoretical (DFT-B3LYP/N07D) spectrum of rosmarinic acid. The contributions into the theoretical (DFT-B3LYP/N07D) NIR spectrum of rosmarinic acid stemming from each calculated band and the final theoretical spectrum. Raw unscaled theoretical data is presented here. Source: Kirchler et al.6—reproduced by permission of The Royal Society of Chemistry.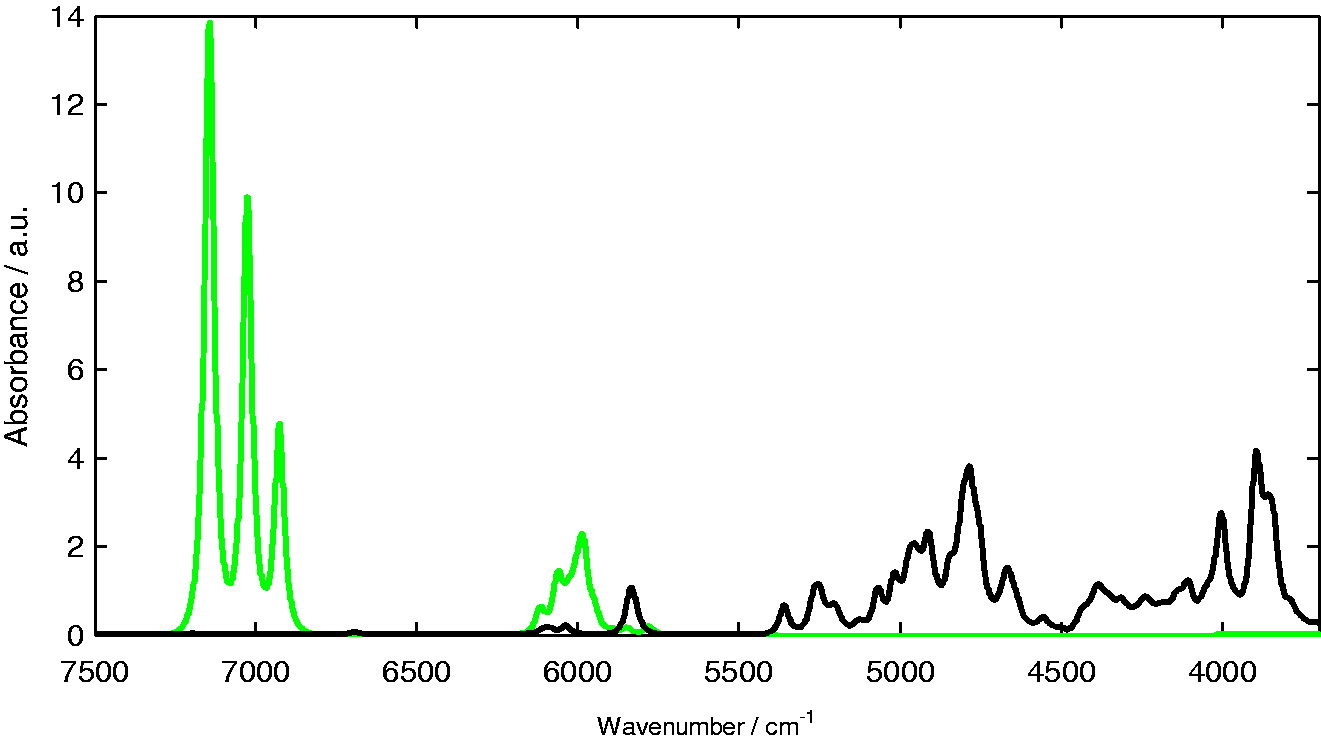
Intensities of binary combination bands appearing in the NIR region of rosmarinic acid.

Note: All values are based on GVPT2 results on DFT-B3LYP/N07D level of theory.
In relation to the most intense NIR binary combination band (above 3700 cm−1).
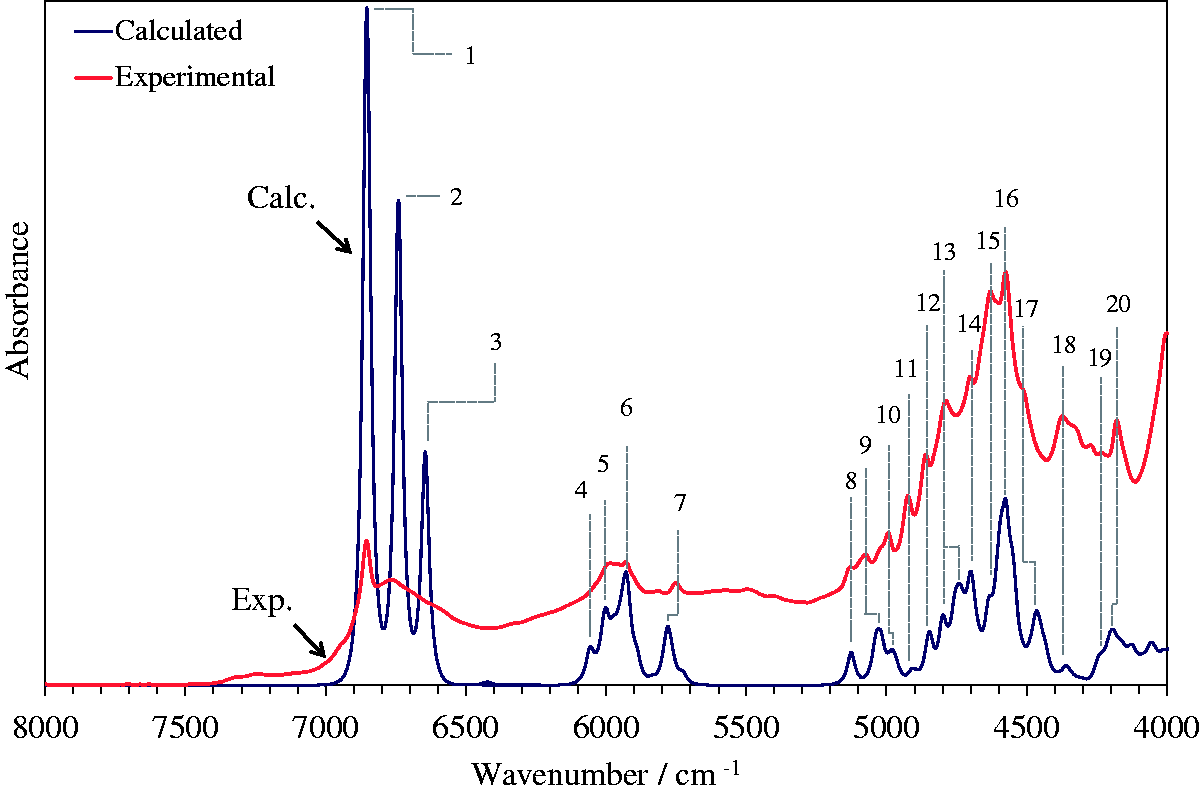
The experimental and theoretical NIR spectrum of rosmarinic acid obtained through fully anharmonic (GVPT2) DFT-B3LYP/N07D calculation. The experimental spectrum is a SNV spectrum normalized over 15 independent experimental datasets. The theoretical bandshapes were obtained with the application of Cauchy-Gauss product function. Most probable assignments of major bands based on calculated spectrum and PED analysis presented in Table 3. Source: Kirchler et al.6—reproduced by permission of The Royal Society of Chemistry.
Intensities of binary combination bands appearing in the NIR region of rosmarinic acid in selected wavenumber regions.
Note: All values are based on GVPT2 results on DFT-B3LYP/N07D level of theory.
In relation to the most intense binary combination band calculated for the entire NIR region (7500 to 3700 cm−1)
Band assignments in NIR spectrum of rosmarinic acid, based on DFT-B3LYP/N07D calculation.

Source: Kirchler et al.6—reproduced by permission of The Royal Society of Chemistry.
Combined approach: 2D-COS, chemometrics and quantum chemistry
The aim of the recent report of Kirchler et al. 6 was a critical evaluation of the analytical performance levels offered by several benchtop and portable NIR devices. The object of the study was R. folium; plant-origin samples were collected on retail phytopharmaceutical market. A detailed overview of analytical performances in quantifying the concentration level of RA in the sample by NIR spectrometers extends beyond the aim of present review. However, we would like to highlight the usefulness of theoretical NIR data in such investigation. NIR spectroscopy often relies on chemometrics in data analysis.2,3 There are also numerous evidences of applications of 2D-COS for analysis of NIR data.2,3,5 In general, among other advantages, these techniques can point out the spectral regions, or individual bands, that correlate with the change of the properties of investigated sample the most. However, the reasons of this correlation, the vibrational modes corresponding to these bands and spectral regions, and thus the conclusions about spectra-structure dependencies were often, out of necessity, presumed and ambiguous. The lack of readily available and reliable source of independent information on the investigated molecular system, often forced NIR applied spectroscopy to rely on data analysis as ‘black-box tool’; at least for complex systems.
Quantum anharmonic methods can provide this independent information, and further allow for a better understanding of the obtained qualitative data. Kirchler et al.
6
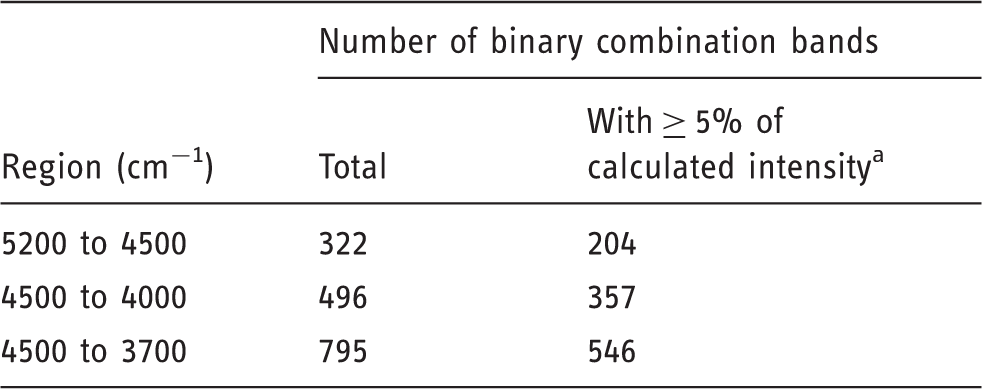
have demonstrated an example of how the results of quantum chemical study can be used as a support of chemometric and 2D-COS plots. They have obtained partial least squares (PLS) regression coefficient plots for the R. folium samples, measured in different and independent experiments using different spectrometers. The example of such PLS data can be seen in Figure 6; here presented for the experimental data obtained on a benchtop NIR device Büchi NIRFlex-N500. A set of wavenumbers of particular interest could be assigned to NIR bands of RA, thanks to the results of quantum chemical calculation.
NIR spectra of Rosmarini folium samples (a) recorded on NIRFlex-N500 benchtop spectrometer and corresponding PLS regression coefficient plot (b). Source: Kirchler et al.6—reproduced by permission of The Royal Society of Chemistry.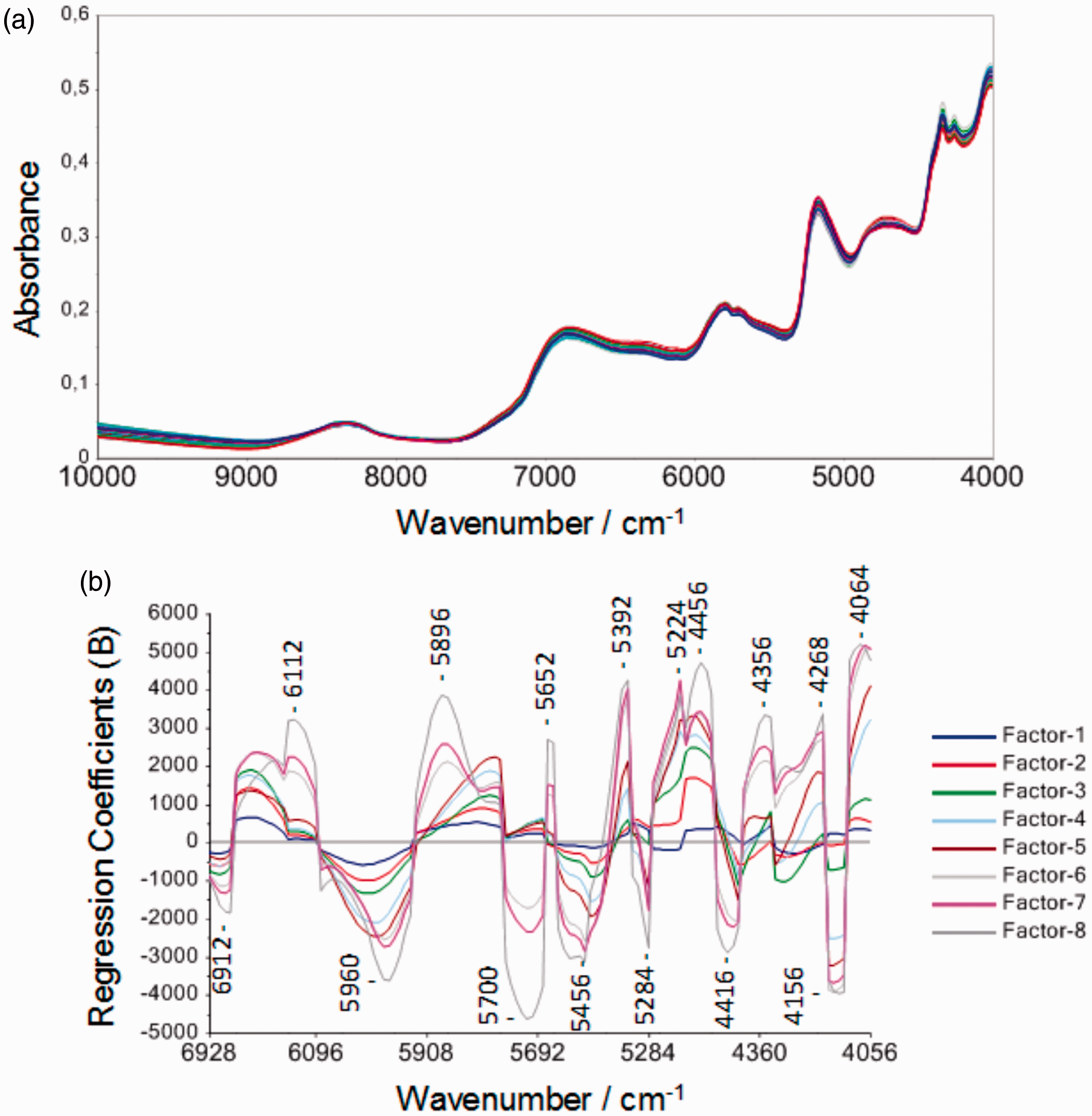
A similar support was provided by theoretical methods in case of hetero-correlation synchronous plots obtained through 2D-COS approach (Figure 7); spectral data originating from different spectrometers was used including Thermo Fisher Phazir and MicroNIR 2200 portable devices. Therefore, a qualitative conclusion about the sensitivity levels offered in different regions by different devices could be made. Theoretical assignments allowed for unambiguous discussion of the observed correlations.
2D-COS synchronous plots of Rosmarini folium samples. (a) Homocorrelation plot for samples measured on NIRFlex N-500 benchtop spectrometer. (b) Homocorrelation plot for samples measured on NIRFlex N-500 benchtop spectrometer versus MicroNIR 2200 portable spectrometer. Source: Kirchler et al.6—reproduced by permission of The Royal Society of Chemistry.
The overviewed investigation performed by Kirchler et al. 6 brought significant novelty on multiple levels into the field of applied NIR spectroscopy. It was the first report which used a quantum chemical NIR spectrum of a complex molecule, investigated analytical performances of portable devices and applied 2D-COS heterocorrelation to elucidate subtle differences in the datasets obtained on different spectrometers. All these multi-level data were combined together, and significant support was provided by quantum chemical calculation. This was an initial novel report, and will certainly be followed by similar investigations. In the future, we should expect that this path will be further explored, using the significant advantages brought by constantly advancing quantum mechanical methods to the applied NIR spectroscopy.
Summary and the perspective for future advances
In the third part of the review series on the quantum chemical methods in NIR spectroscopy, we briefly introduced to the topic and discussed the main aspects of applying anharmonic quantum methods to complex molecules. On the basis of recent reports in the field, we demonstrated that modern quantum chemistry allows for an accurate reproduction of NIR spectra of relatively large molecules. On the example of theoretical data obtained for RA we demonstrated how NIR spectrum is formed, and we presented the reasons of inherent complexity of NIR spectra.
In the past, the basic studies in NIR often focused on overtone bands; one of the major reasons was that investigation of overtones is less challenging, as one can use the corresponding fundamental bands as an additional source of information. The fundamental bands can be calculated using the routine harmonic quantum methods, and therefore the support for the analysis of overtone bands was readily available. However, the investigation of RA molecule evidenced, that the major role in forming NIR spectrum of organic molecules is played by combination modes. As the analysis of these requires the use of fully anharmonic approaches, no clear evidence of such studies applied to complex molecules was ever reported before. One can expect, that recent advances in theoretical methods will allow to explore this topic further.
We anticipate that the importance of anharmonic quantum mechanical studies applied to complex molecules will grow significantly in the forthcoming years. This kind of systems form the most typical object of interest for applied NIR spectroscopy; this concerns material science, biochemistry, pharmaceutical chemistry, and particularly, phytopharmaceutical chemistry. Complex molecules are also typical for natural products, which in the nearest future should be gaining even more attention from multiple directions from the analytical science and industry. In all of these fields, which often rely on NIR spectroscopy and advanced data analysis methods, the advantages stemming from obtaining a powerful and independent source of information, the anharmonic calculations, can be extremely helpful – as in case of the overviewed RA study.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.