
Abstract
Thrombosis is a frequent manifestation in patients with systemic lupus erythematosus (SLE), although precise mechanisms remain unclear. This study investigated whether the major physiological trigger of blood coagulation, the tissue factor (TF) pathway, was altered in SLE patients. Furthermore, we investigated potential associations between the TF pathway, the presence of antiphospholipid (APL) antibodies and other abnormalities present in SLE. A total of 101 participants (40 SLE patients and 61 age- and sex-matched controls) were recruited from Tasmania, Australia. Markers of the TF pathway, hypercoagulability, inflammation and endothelial cell damage were measured in plasma. Serum levels of APL antibodies (anti-cardiolipin antibodies [ACL], lupus anticoagulants [LAC], anti-beta2-glycoprotein-1 [anti-β2GP1] and anti-prothrombin antibodies) were also determined. Despite similar TF and TF pathway inhibitor (TFPI) total antigen levels, SLE patients had significantly increased levels of TFPI free antigen (patients vs controls; mean ± SD) (11.6 ± 0.9 ng/mL vs 6.4 ± 0.4 ng/mL; p < 0.001) but significantly reduced TFPI activity (0.66 ± 0.07 U/mL vs 1.22 ± 0.03 U/mL; p < 0.001), compared with healthy controls. Anti-TFPI activity, designated as the ability of isolated IgG fractions to inhibit TFPI activity in normal plasma, was detected in 19/40 (47.5%) of SLE patients and 3/40 (7.5%) of healthy controls. The significant reduction in TFPI activity in SLE patients reflects impaired functional control of the TF pathway. Moreover, SLE patients with a history of thrombosis demonstrated higher levels of TFPI activity compared with patients without a previous thrombotic event (0.97 ± 0.07 U/mL vs 0.53 ± 0.14 U/mL; p = 0.0026). Changes to the TF pathway were not associated with manifestations of SLE such as inflammation or endothelial cell damage. The results from this study suggest hypercoagulability in SLE may (in part) be due to reduced TFPI activity, a mechanism that appears to be independent of other abnormalities in SLE.
Introduction
SLE is a chronic inflammatory disease that can affect any part of the body, including the skin, joints, liver, kidneys and blood. Haematological complications are common in SLE, including anaemia, the presence of circulating APL antibodies (e.g. ACL, LAC, and anti-β2GP1 antibodies) and thrombotic manifestations. Indeed, it has been reported that patients with SLE who are positive for APL antibodies are three times more likely to have a thrombotic event compared with subjects who are negative for APL antibodies, 1 although the precise mechanisms are yet to be fully elucidated. 2 Other haemostatic factors, including Factor V Leiden genotype, 3 platelet activation4,5 and the action of APL antibodies on monocytes to induce tissue factor (TF) expression and procoagulant activity, 6 – 8 have also been proposed to contribute to hypercoagulability in SLE patients.
TF is an integral membrane protein that is not normally expressed on cells in contact with circulating blood. Following vessel injury TF complexes with coagulation factor VII(a) to initiate a series of protein/enzyme interactions through the TF pathway, resulting in thrombin generation, coagulation amplification, platelet activation, and fibrin deposition. TF is regulated by tissue factor pathway inhibitor (TFPI), a Kunitz-type protease inhibitor that is well described as the major regulator of the TF pathway. 9 – 12 TFPI acts in a factor Xa-dependent manner to inhibit the TF-factor VIIa complex, and thus regulate thrombin generation and fibrin formation. Although varying circulating levels of TFPI have been reported in many diseases, a ‘TFPI deficiency’ phenotype has yet to be clearly defined.
Altered balance of the TF pathway, usually through increased TF expression that is not compensated by a similar increase in TFPI, is thought to contribute to increased hypercoagulability in a variety of conditions including sepsis, 13 acute respiratory distress, 14 disseminated intravascular coagulation, 15 and the antiphospholipid syndrome, 10 but has yet to be extensively investigated in patients with SLE. The aim of the present study was to therefore investigate whether there were changes to the TF pathway in a well-defined cohort of SLE patients, compared with healthy controls. Furthermore, we investigated potential associations between markers of the TF pathway, and the presence of different APL antibodies, and other abnormalities present in SLE (i.e. inflammation and endothelial cell damage).
Materials and methods
Controls and patients
This study was approved by the Tasmanian Health and Medical Human Research Ethics Committee (Reference number: H009415). A total of 101 subjects were recruited, comprising 40 SLE patients and 61 age- and sex-matched healthy controls. SLE patients met the American College of Rheumatology Classification, 16 and time (mean ± SD) since diagnosis was 10.1 ± 7.3 years. All participants were recruited through the University of Tasmania, the Lupus Association of Tasmania and the Launceston General Hospital (Tasmania), and provided informed consent.
Approximately one-third of SLE patients (n = 12) reported a previous thrombotic event. These included lower limb deep vein thrombosis (DVT) (n = 8), thrombosis in the groin (n = 2), ovary (n = 2) and knee (n = 1), pulmonary embolism (n = 3), stroke (n = 2), post-operative DVT (n = 3), post-partum DVT (n = 2), miscarriage (n = 1) and thrombophlebitis (n = 1). More than one type of thrombotic episode was reported by six (16%) SLE patients. No healthy control reported a previous thrombotic event.
Blood sampling
Whole blood (approximately 6 mL) was collected from the cubital vein. For the detection of APL antibodies, blood was collected into tubes without anticoagulant to obtain serum. For the measurement of markers of the TF pathway, thrombin generation, inflammation and endothelial cell damage, one part whole blood was collected into nine parts sodium citrate (3.8%) and centrifuged at 2000 g for 10 minutes. The plasma was separated and centrifuged a second time at 2000 g for 10 minutes to obtain platelet poor plasma (PPP). Whole blood was collected from a further 20 healthy subjects to obtain pooled normal plasma (PNP) and used to generate standard curves for the TFPI and anti-TFPI activity assays. All samples were stored in 1 mL aliquots at −80°C until assayed.
Isolation of IgG fractions
IgG fractions were isolated from PPP by affinity purification using protein G columns (GE Healthcare, Uppsala, Sweden). Samples were diluted 1:5 in 0.02 mol/L phosphate buffered saline (PBS), pH 7.4. Prior to use, columns were washed and equilibrated with 10 mL of PBS. One hundred µL 1 M Tris-HCl (pH 9.0) was added to collection tubes to neutralize the pH of the fraction collected. A 5 mL diluted sample was applied to the column, which was then washed with 5 mL of PBS. IgG was eluted from the column using 5 mL glycine, pH 2.7. The purified fractions were buffer exchanged with PBS using a desalting column (GE Healthcare, Buckingham, UK). After elution, the absorbance of each 1 mL fraction was measured at 280 nm, and the aliquot with the highest absorbance was added to 1.5 mL PBS pH 7.4. This volume was applied to the desalting column that was equilibrated using 25 mL PBS, pH 7.4. The flow through was discarded, then IgG fractions were eluted using 3.5 mL buffer. Equal volumes of each fraction were pooled and stored at -80°C until required.
Antiphospholipid antibodies
Serum samples were analysed using commercially available assays to determine levels of APL antibodies. The assays were; Staclot® dRVV LA Detection Kit for lupus anticoagulants (Diagnostica Stago, Asnieres, France), enzyme linked immunosorbent assay (ELISA) for IgG and IgM isotypes of ACL (Enzyme LinkedImmuno Concepts®, Sacramento, USA), and ELISAs for anti-β2GP1 and anti-prothrombin IgG antibodies (Corgenix, Inc., Broomfield, USA). All assays were performed according to manufacturer’s instructions.
Laboratory methods
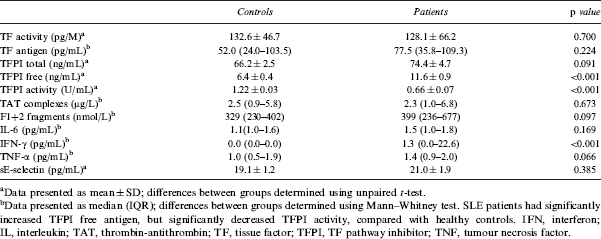
Markers of the TF pathway, thrombin generation, inflammation and endothelial cell damage were measured using commercially available assays according to manufacturer’s instructions, unless otherwise stated. TF, TFPI total antigen and TFPI free antigen were measured using ELISA (American Diagnostica Incorporated, Greenwich, USA). TF activity was measured using a chromogenic activity assay (Abnova, Taipei City, Taiwan). Thrombin-antithrombin (TAT) complexes and prothrombin fragment 1+2 (F1+2 fragments) were measured using ELISA (Dade-Behring, Marburg, Germany). Interleukin 6 (IL-6) was measured using a high sensitivity quantitative enzyme immunoassay (Quantikine1 HS; R&D Systems Inc., Minneapolis, USA). Human soluble E-selectin (sE-selectin) was measured by ELISA (Hycult Biotechnology b.v., Uden, The Netherlands).
TFPI activity was measured using an amidolytic assay as previously described. 17 – 19 Anti-TFPI activity was determined using a slight modification of the TFPI activity assay. Equal volumes of IgG fractions (25 µg/mL) isolated from SLE patients and healthy controls were mixed with PNP (containing 1 U/mL TFPI activity) for 30 minutes at 37°C. To determine anti-TFPI activity, the TFPI activity of PNP with IgG was subtracted from the TFPI activity of PNP mixed with TFPI activity buffer (standard) to demonstrate the effect of IgG on TFPI activity.
Statistical analysis
Statistical analysis was performed using Statistical Package for Social Sciences (SPSS v18, Chicago, USA). The data is presented as mean ± standard deviation (SD) for normally distributed data, or as median (interquartile range; IQR) for non-normally distributed data. Differences between groups were determined using either an unpaired t-test or Mann–Whitney test, depending on the distribution of data. Associations between the various laboratory markers were determined using Pearson’s correlation coefficient (r) or Spearman's rank correlation coefficient (rho), depending on the distribution of data. P-values <0.05 were considered statistically significant.
Results
Controls and patients
Demographic and APL antibody data of healthy controls and SLE patients
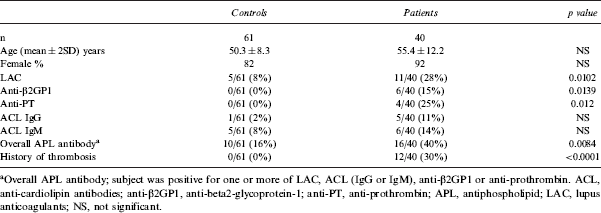
Overall APL antibody; subject was positive for one or more of LAC, ACL (IgG or IgM), anti-β2GP1 or anti-prothrombin. ACL, anti-cardiolipin antibodies; anti-β2GP1, anti-beta2-glycoprotein-1; anti-PT, anti-prothrombin; APL, antiphospholipid; LAC, lupus anticoagulants; NS, not significant.
Markers of the TF pathway
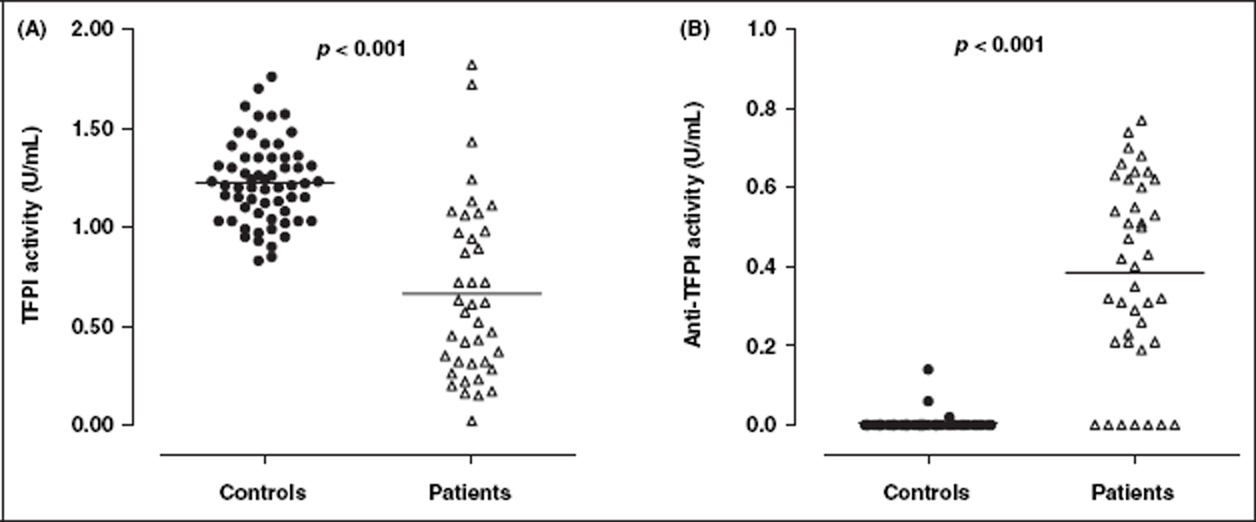
Results are summarized in Table 2. The median level of TF antigen in SLE patients was increased compared with healthy controls, but this was not statistically significant (77.5 vs 52.0 pg/mL; p = 0.224) (Figure 1A). Plasma levels of TFPI antigen (total) were not statistically different between controls and patients (Figure 1B). SLE patients had higher levels of TFPI antigen (free) (mean ± SD) (11.6 ± 0.9 vs 6.4 ± 0.4 ng/mL; p < 0.001) (Figure 1C), but lower mean TFPI activity (0.66 ± 0.07 vs 1.22 ± 0.03 U/mL; p < 0.001) (Figure 2A), compared with healthy controls. Anti-TFPI activity was significantly higher in SLE patients compared with healthy controls (Figure 2B), and was detected in 19/40 (47.5%) of patients and 3/40 (7.5%) of controls tested.
Plasma levels of TF and TFPI in healthy controls and SLE patients. Although plasma levels of both TF antigen (A), TF activity (B) and TFPI total antigen (D) were not significantly different, SLE patients (n = 40) had significantly increased levels of TFPI free antigen (C) compared with healthy controls (n = 61). Solid horizontal lines indicate mean values for TF activity, TFPI total antigen and TFPI free antigen, and median values for TF antigen. TF, tissue factor; TFPI, TF pathway inhibitor. TFPI and anti-TFPI activity in healthy controls and SLE patients. SLE patients (n = 40) had significantly reduced levels of TFPI activity (A) i.e. function, but increased levels of anti-TFPI activity (B) compared with healthy controls (n = 61). Solid horizontal lines indicate mean values. TFPI, TF pathway inhibitor. Markers of the TF pathway, thrombin generation, inflammation and endothelial cell damage in healthy controls and SLE patients Data presented as mean ± SD; differences between groups determined using unpaired t-test. Data presented as median (IQR); differences between groups determined using Mann–Whitney test. SLE patients had significantly increased TFPI free antigen, but significantly decreased TFPI activity, compared with healthy controls. IFN, interferon; IL, interleukin; TAT, thrombin-antithrombin; TF, tissue factor; TFPI, TF pathway inhibitor; TNF, tumour necrosis factor.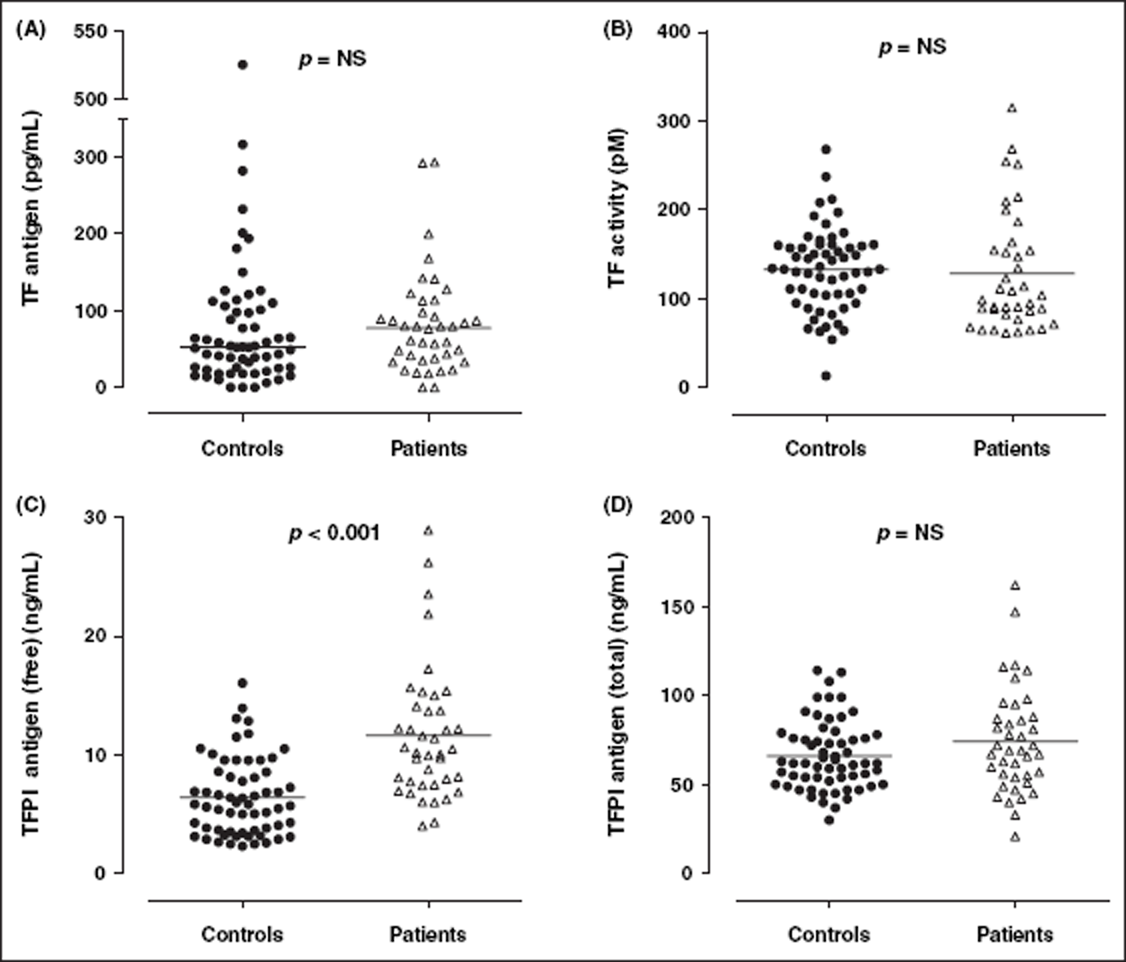
Analysis of TF:TFPI ratio
To further evaluate changes to the balance of the TF pathway, the ratio of TF to various measures of TFPI were determined in healthy controls and SLE patients (Figure 3). There was no significant difference in the ratio of TF(antigen): TFPI(total antigen), (controls vs patients; median[IQR]) (0.71 [0.34–1.60] vs 1.00 [0.50–1.50]; p = 0.42) or TF(antigen):TFPI(free antigen) (9.1 [3.0–19.9] vs 6.6 [3.5–9.2]; p = 0.054). However, there was a statistically significant difference in the ratio of TF(antigen): TFPI(activity) (42.1 [18.5–95.2] vs 99.0 [63.4–281.4]; p < 0.001) and TF(activity): TFPI(activity) (112.5 [77.1–135.9] vs 201.5 [105.50–407.7]; p < 0.001), between the two cohorts (Figure 3).
Ratio of TF:TFPI. There was no significant difference in the ratio of TF antigen to either TFPI total (A) or TFPI free antigen (B) between SLE patients (n = 40) and healthy controls (n = 61). However, SLE patients had a significantly increased ratio of TF antigen to TFPI activity (C) and TF activity to TFPI activity (D), reflecting impaired control of the TF pathway of blood coagulation. Solid horizontal lines indicate median values. TF, tissue factor; TFPI, TF pathway inhibitor.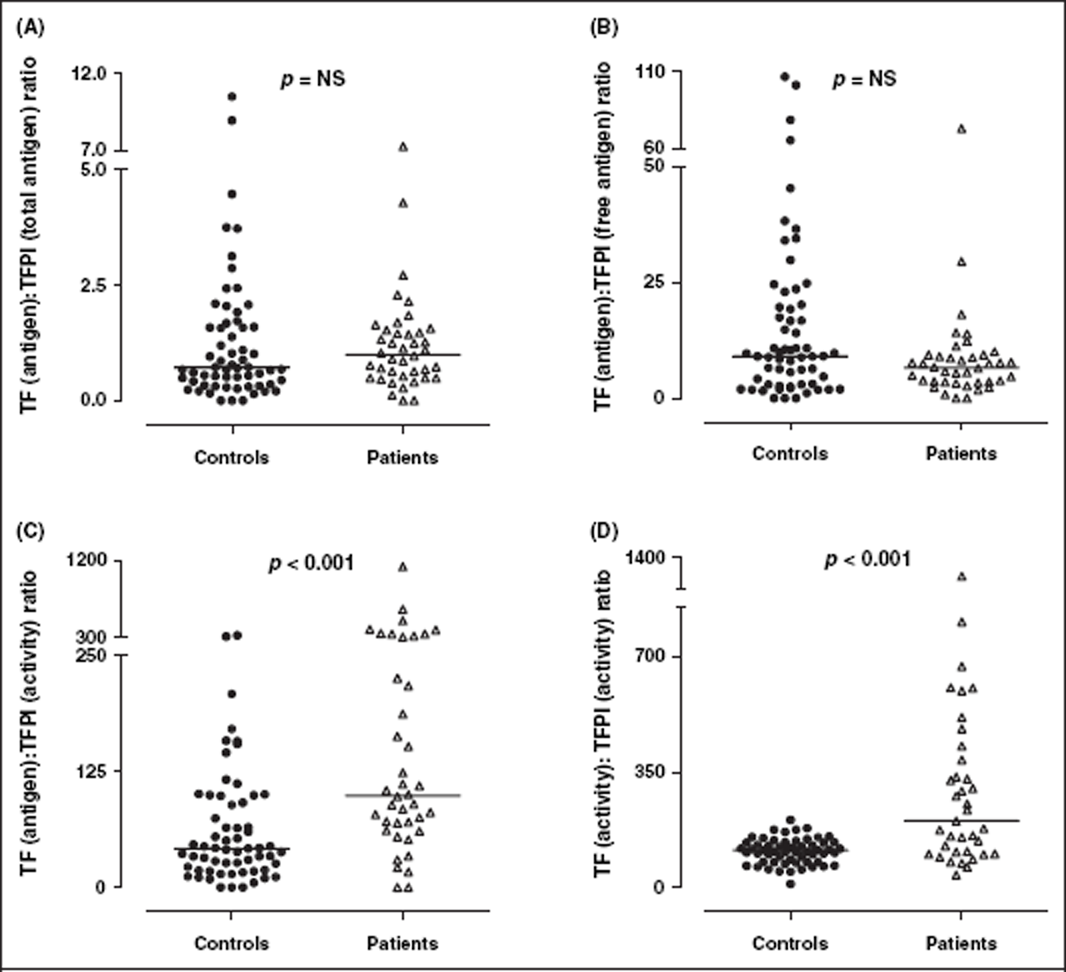
Markers of thrombin generation, inflammation and endothelial cell damage
SLE patients had significantly higher levels of interferon-gamma (IFN-γ) compared to healthy controls. SLE patients had higher levels of F1+2 fragments than controls, but this was not statistically significant (p = 0.091). There was no significant difference in the level of TAT complexes in SLE patients and healthy controls. Similarly, there were no significant differences in laboratory markers of inflammation (IL-6, tumour necrosis factor alpha [TNF-α]) or endothelial cell damage (sE-selectin) between patients and controls. Results are summarized in Table 2.
Correlation analysis
TFPI free antigen was significantly associated with TFPI total antigen in both healthy controls (r = 0.348; p = 0.006) and SLE patients (r = 0.734; p < 0.001) (Figure 4). However, TFPI activity was not associated with TFPI total or free antigen in either controls or patients (both p > 0.05). Furthermore, there was no association between TFPI activity and anti-TFPI activity in either controls or patients (p > 0.05).
Association between TFPI total and free antigen levels in healthy controls (•) and SLE patients (x). There were significant, positive associations between TFPI total and free antigen levels in both healthy controls (r = 0.348; p = 0.006) and SLE patients (r = 0.734; p < 0.001), however TFPI activity was not associated with TFPI total or free antigen levels, in either controls or patients. TFPI, TF pathway inhibitor.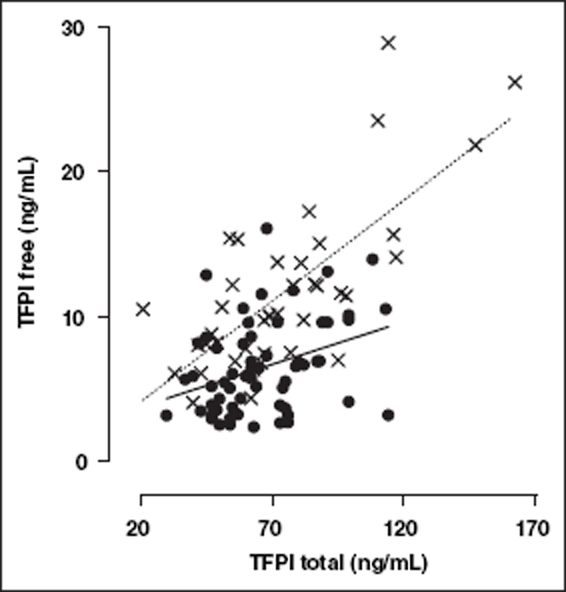
In healthy controls, there were significant associations between TFPI activity and ACL IgG (r = −0.355; p = 0.005), anti-β2GP1 and TFPI free antigen (r = 0.395; p = 0.002), anti-prothrombin antibodies and TFPI free antigen (rho = 0.439; p < 0.001), F1 + 2 and TFPI free antigen (rho = 0.471; p < 0.001), and IL-6 and TFPI free antigen (rho = 0.411; p = 0.001). In SLE patients, there were no significant associations between the parameters measured.
TFPI activity was significantly elevated in SLE patients with a history of thrombosis, compared with patients who had not experienced a thrombotic event (n = 12 (0.97 ± 0.07 U/mL) vs n = 28 (0.53 ± 0.14 U/mL); p = 0.0026) (Figure 5). Furthermore, there were no significant differences between SLE patients according to APL status i.e. APL negative vs SLE APL positive, for any of the measured parameters (data not shown).
TFPI and anti-TFPI levels in SLE patients (grouped according to history of thrombosis). TFPI activity was significantly elevated in SLE patients with a history of thrombosis. There was no significant difference in TFPI antigen (total or free), or anti-TFPI activity, in SLE patients with a history of thrombosis compared with those without history of thrombotic event. Solid horizontal lines indicate mean values. TFPI, TF pathway inhibitor.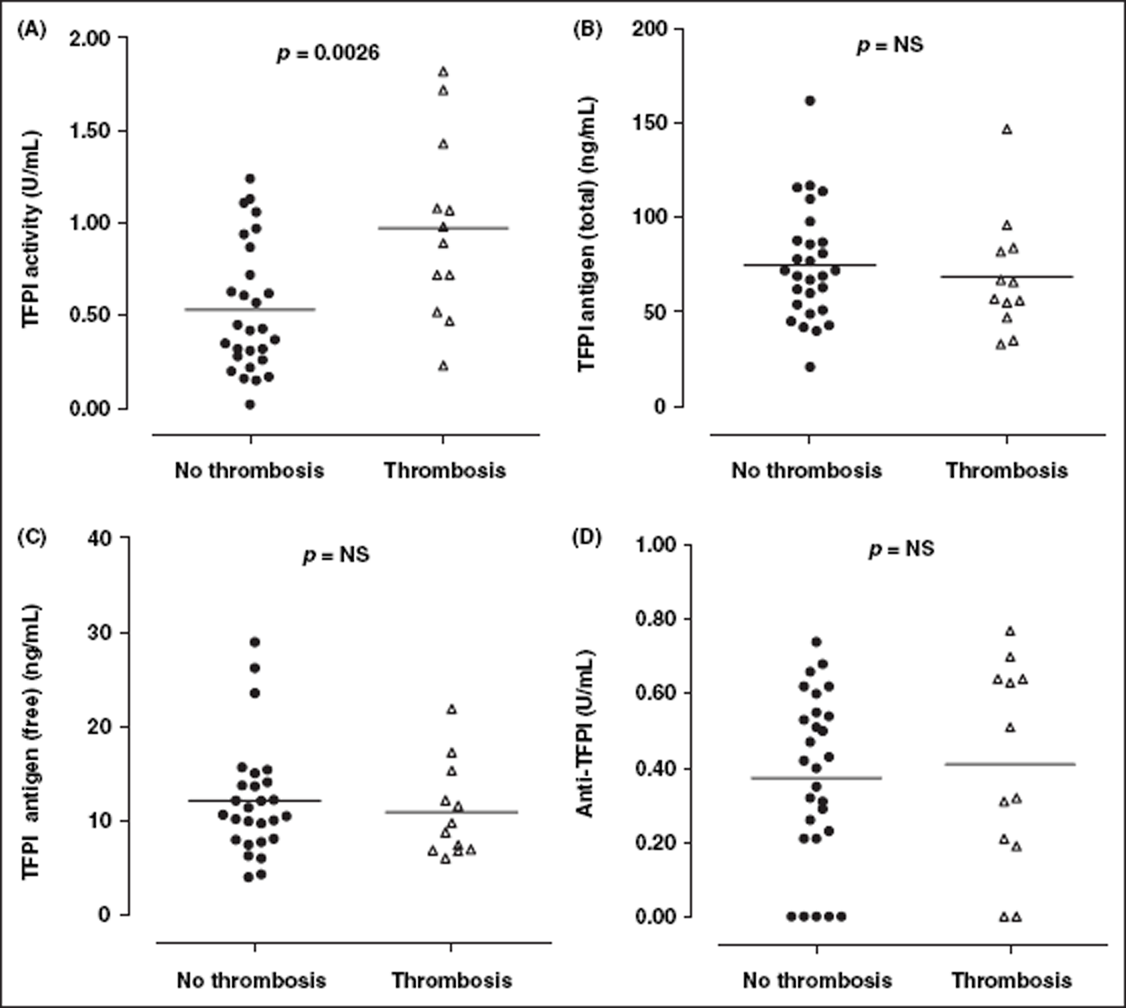
Discussion
The present study has demonstrated impaired TFPI regulation of the TF pathway of blood coagulation in patients with SLE, compared with healthy controls. Interestingly, this was not reflected by changes in either TF (antigen or activity) or TFPI total antigen levels, but was rather due to significantly reduced plasma levels of TFPI activity in SLE patients. Furthermore, we demonstrated that IgG fractions isolated from SLE patients significantly inhibit TFPI activity of normal pooled plasma. Anti-TFPI activity was detected in approximately half of the IgG fractions isolated from SLE patients in this study cohort, although it was not associated with history of thrombosis. Moreover, changes to the regulation of the TF pathway were not associated with inflammation or endothelial cell damage, suggesting that the development of hypercoagulability is independent of these clinical manifestations in SLE.
SLE patients demonstrated higher levels of APL antibodies (LAC, anti-β2GP1 and anti-prothrombin) compared with healthy controls, consistent with other studies.20,21 It has previously been demonstrated that approximately 50% of SLE patients and 1–5% of the general population are positive for APL antibodies. 22 – 24 The heterogeneity and transient nature of APL,25,26 as well as the lack of standardized diagnostic methods, contribute to some difficulty in detecting the presence of APL, 27 which may also help explain the lack of association between APL and markers of the TF pathway. Moreover, APL antibody levels increase in patients with chronic disease and older individuals, 23 and together with the relatively small sample size of the current study, may explain the higher incidence of APL antibodies demonstrated in healthy controls of the present study (16%), where the mean age was 50 years.
There is minimal information available in the literature about the role of the TF pathway in SLE. In this study, we have demonstrated no significant difference in TF antigen levels between SLE patients and healthy controls. It was somewhat unexpected that increased levels of TF were not demonstrated as it has previously been reported that monocytes from SLE patients express increased levels of cell surface TF, particularly in patients who had a previous thrombotic event. 28 In the current study we hypothesized that higher circulating TF would reflect increased expression due to an ongoing inflammatory state and endothelial cell disruption. It may be that TF expressed by monocytes has a more significant role in modulating the ongoing thrombin generation in SLE, compared with that expressed by endothelial cells. Our data provides some support for this statement in that sE-selectin levels in SLE patients were similar to healthy controls, suggesting minimal or no endothelial cell disruption, although this may be confounded by immunosuppressive treatment.
Our data demonstrates no significant changes to TFPI total antigen levels between the patient and control groups. This is in contrast to previous studies that have reported both increased 29 and decreased 30 TFPI total antigen levels. The reasons for the varying results are unknown, however, increased TFPI total antigen may reflect endothelial cell injury. Again, sE-selectin levels were not altered in our study, which may in part provide an explanation of the similar patient and control TFPI total antigen levels. Decreased circulating TFPI total antigen levels have also previously been reported, however the vascular endothelial pool of TFPI was not affected following heparin administration. 30
Free TFPI is the 43 kDa full length form of the inhibitor that circulates in plasma, but is not associated with lipoproteins. It is thought to play a more biologically active role in regulating the TF pathway as it has greater anticoagulant activity than the lipoprotein bound forms of TFPI.31,32 The increase in TFPI free antigen levels in SLE patients reported in the current study is in agreement with earlier work, 29 and may reflect either increased release from intracellular/cell surface-associated stores, endothelial cell injury, 29 release from platelets following activation, 33 or possibly redistribution from (primarily) low density lipoprotein stores.
A limitation of many studies that investigate the role of the TF pathway in normal haemostasis and disease is that they do not assess the activity of TFPI using functional assays. These techniques more appropriately reflect the in vivo situation compared with antigen methods, as their principle depends on the ability of TFPI to inhibit TF-FVIIa-FXa (the complex formed following the initiation of coagulation). To our knowledge, the current study is the first to report TFPI activity in a well-defined cohort of SLE patients, compared with healthy controls.
We demonstrated that TFPI activity was decreased in SLE patients despite normal TFPI total antigen and increased TFPI free antigen levels. These results suggest that although TFPI, particularly the more biologically active free form, is present at normal to increased levels in SLE patients, it does not have the same inhibitory capacity as TFPI in normal plasma. Furthermore, in SLE patients with a history of thrombosis, TFPI activity levels were unexpectedly increased, compared with patients who had not previously had a thrombotic event. Reduced circulating TFPI activity levels in SLE patients may therefore reflect a pre-thrombotic environment and contribute to the development of thrombosis in conjunction with other risk factors.
It is interesting to speculate why the SLE cohort in this study demonstrated reduced TFPI activity. Possible reasons may include the action of serine proteases such as plasmin, thrombin and neutrophil elastase, 34 – 36 that have previously been demonstrated to cleave TFPI, possibly causing reduced anticoagulant function. Other possibilities may include the presence of autoantibodies and/or cross-reacting APL antibodies that partially or completely attenuate TFPI function. The current study has also confirmed the presence of an inhibitory component to TFPI in approximately half (48%) of the IgG fractions isolated from the plasma of SLE patients. This is in agreement with earlier studies using samples obtained from patients with the antiphospholipid syndrome, which found 65% 17 and 48%. 19 Our data suggests that the inhibitory component in IgG fractions from SLE patients is unlikely to be an APL antibody, as there was no clear association between any of the APL antibodies studied and anti-TFPI activity. Moreover, it is noted that the majority of the SLE cohort in this study was receiving immunosuppressive agents which would, 1) suggest that the mechanism of impaired control of the TF pathway is independent of inflammation, 2) reduce the likelihood of immune production of autoantibodies, and 3) provide a reason why IL-6 and IFN-γ levels were not significantly increased, compared with healthy controls.
It is acknowledged that the current study has two important limitations. Firstly, there was a relatively small sample size to detect significant differences and/or potential associations between the markers investigated. Secondly, methods used to measure TFPI demonstrate variable correlation when comparing results obtained using functional (activity) and protein (antigenic) techniques, 37 – 39 which is due to the different principles of detection used by these methods. ‘Activity’ assays measure the functional capacity of TFPI to inhibit TF-FVIIa-FXa complexes, whereas ‘antigenic’ methods use antibodies to capture different regions of the TFPI molecule, and thus detect the overall protein level of TFPI. Moreover, the plasma pool of TFPI is a heterogeneous mixture of different molecular weight forms of the inhibitor that contains variable functional capacity due to the association with lipoproteins and the probable degradation of the molecule by plasma proteases. Therefore, it was not surprising that the data in the current study demonstrated good correlation when comparing total and free antigen levels of TFPI (Figure 5), but not when comparing antigen with activity. This confirms the importance of determining the functional ability, rather than just the presence of the protein, in studies investigating TFPI.
In conclusion, this study has demonstrated that despite normal protein levels of TFPI, SLE patients have both decreased TFPI activity and increased anti-TFPI activity, compared with healthy controls. These changes reflect impaired regulation of the TF pathway that may contribute to hypercoagulability in SLE. Furthermore, these changes were not associated with other markers that reflect clinical manifestations of SLE, i.e. inflammation and endothelial cell damage. Further studies are clearly warranted to determine how TFPI activity is impaired, and whether there are other co-factors involved in this interference to the TF pathway.
Footnotes
Acknowledgements
The authors would like to thank the Lupus Association of Tasmania for their support of this study. We are also grateful for the cooperation of everyone at Launceston Pathology, Hobart Pathology, North-West Pathology (Burnie and Latrobe) and the Pathology Department at the Launceston General Hospital in helping with sample collection and processing. We would also like to thank Mrs Merrilyn Johnson and Mrs Natasha Betts for their excellent technical assistance.
Funding
This work was supported by a grant from the Clifford Craig Medical Research Trust (Project 66), and a Faculty of Health Science Strategic Grant, University of Tasmania.