
Abstract
Keywords
Introduction
Systemic lupus erythematosus (SLE) is a multi-system, chronic autoimmune disorder. The clinical course of SLE is diverse. Hematological involvement is of significant importance in SLE in terms of morbidity and mortality. Hematological findings may be present at the onset of the disease or develop during the course. In previous studies, hematological involvement has been reported to range between 34% and 79%.1,2 Anemia, especially Coombs positive hemolytic anemia, is the most common hematological finding. 2
We have aimed to describe the hematological features and their implications in childhood SLE. We have also reviewed our treatment strategies and outcomes for these patients.
Patients and methods
In this descriptive case series, we analyzed the hematological findings of 43 children with SLE diagnosed and followed up at the Department of Pediatric Rheumatology at Hacettepe University, Ankara, Turkey, in the last 10 years. The associations of hematological abnormalities with autoantibodies and other organ involvement were also reviewed.
All patients in this study fulfilled the ACR classification criteria for SLE. 3 Patients with drug-induced lupus, overlap syndrome and mixed connective tissue disorders were excluded from the study.
All patients were reviewed in detail for hematological findings, including complete blood count, Coombs test, coagulation parameters and bone marrow analysis — if performed. Antiphospholipid and anticardiolipin antibodies were analyzed in 38 patients. Thrombocytopenia (platelet ≤ 150 × 109/L), leucopenia (white blood cell ≤ 4 × 109/L) and neutropenia (absolute neutrophil count ≤ 1.5 × 109/L) were evaluated in all.
Anemia was defined when the hemoglobin level was 2 SD below the normal hemoglobin level for age. Chronic anemia of inflammation, iron deficiency anemia and hemolytic anemia were diagnosed according to the results of serum iron, iron binding capacity, ferritin, Coombs test and reticulocyte count.
Macrophage activation syndrome (MAS) was diagnosed when five of the eight criteria of hemophagocytic lymphohistiocytosis (HLH) of the Histiocyte Society were fulfilled. The HLH-2004 treatment protocol consisted of cyclosporine, dexamethasone, etoposide and intravenous gamma globulin (IVIG). 4
Antiphospholipid and anticardiolipin antibodies (IgM and IgG) were assayed using a standard ELISA method.
Anti-nuclear antibody (ANA) and anti-dsDNA antibody were determined by indirect immunoflourescence using Hep-2 cells and ELISA, respectively.
All information was saved and the confidentiality of the patients was ascertained.
Simple descriptive analysis including mean, median and range was used to summarize the results with the help of SPSS 10.0 for Windows.
Results
The median age of 43 patients (35 females) at presentation was 13 years (range, 4–17 years) and median follow-up was 26 months (range, 5–158 months). The most common clinical features were articular involvement (93%), followed by hematological (86%) and renal manifestations (62.8%) in this cohort. Central nervous system involvement was noted in five patients (11.6%). ANA and anti-dsDNA were positive in 42 (97.6%) and 36 (83.7%) patients, respectively.
Overall, 86% (n = 37) of the SLE patients had hematological abnormalities. Of these, 16 patients (43.2%) displayed hematological findings at presentation. Almost half (n = 9) presented with autoimmune hemolytic anemia (AIHA), four with thrombocytopenia and one with thrombosis. An additional two patients — without cytopenia— who had presented with arthralgia and weight loss were referred initially to the pediatric hematology clinic in order to rule out hematologic malignancy.
Anemia and cytopenia
Evaluation of probable causes of cytopenias in our cohort
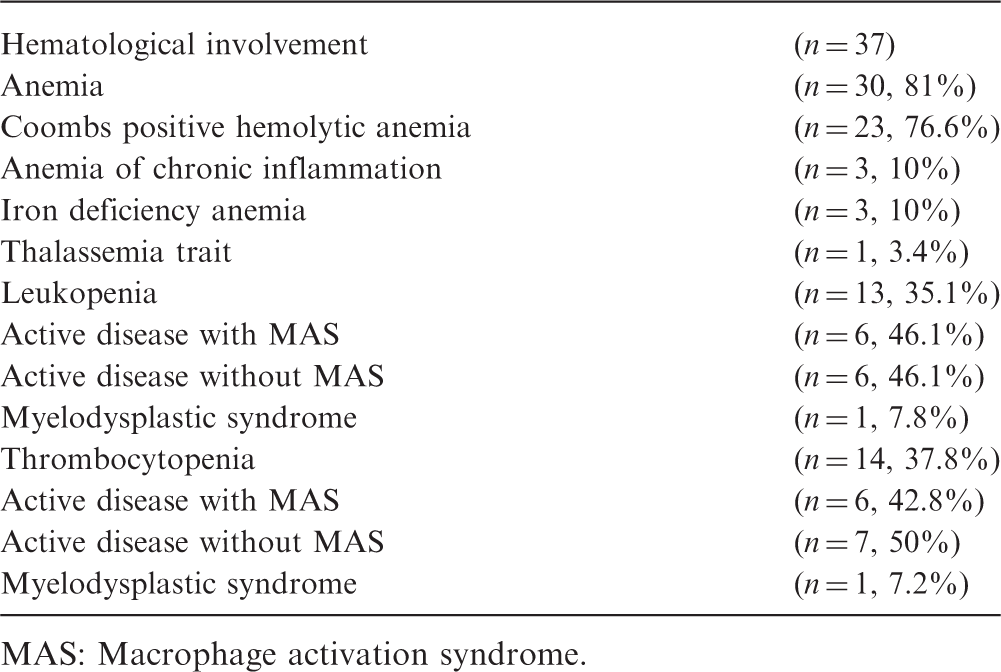
MAS: Macrophage activation syndrome.
Treatment
All of these patients were treated with steroid pulses and/or IVIG. We have used IVIG in 12 of our patients with hemolytic anemia with a persistent response in nine of them. We used rituximab (375 mg/m2, per week) in three patients for resistant hemolytic anemia. Coombs test became negative and the hemoglobin level increased. Subsequent to the return of cell counts to normal, these three latter patients who also had class IV nephritis received cyclophosphamide without further hematological complications. Granulocyte-colony stimulating factor (G-CSF) was administered at a dose of 10 µg/kg/day to three of seven neutropenic patients, with an excellent response.
Myelodysplasia and leukemia
Bone marrow aspiration was performed in 15 (40.5%) patients mainly to assess the cytopenia. Secondary dysplastic changes in myeloid and erythroid lineage, increased histiocytes, megaloblastic changes and erythroid lineage hyperactivity were common findings. In one of the patients, myelodysplastic syndrome subsequently became apparent, and acute lymphoblastic leukemia (ALL) was diagnosed at the age of 6 years, nine months after the diagnosis of SLE. She had been treated with corticosteroids and azathioprine (AZA) for her SLE over the previous four months.
Treatment
No specific treatment was introduced on detection of myelodysplasia since it was mainly attributed to the disease per se. The child with leukemia was put on St Jude T15 protocol for ALL. 5 During the remission induction phase of the protocol, she unfortunately died due to mucormycosis.
Secondary hemophagocytosis (MAS due to SLE)
Evidence of hemophagocytosis was present in bone marrow smears of six patients. The first three patients had a severe presentation with splenomegaly, fever, marked cytopenia, high ferritin, high triglyceride and low fibrinogen. The latter three patients had ferritin levels above 1000 ng/ml, cytopenia and fever. They had a normal fibrinogen/triglyceride level and no splenomegaly. Although they did not fulfil HLH criteria of the Histiocyte Society, 4 they were also accepted to have MAS.
Treatment
The first three patients with more severe presentations were treated with HLH-2004 protocol, 4 and plasma exchange was performed for all. One of these three patients died of secondary infections and multiorgan failure following a protracted course of HLH. The latter three patients were treated with steroid pulses followed by oral steroids, IVIG and cyclosporine. All three had a quick recovery.
Antiphospholipid syndromes and thrombotic episodes (including purpura fulminans)
Antiphospholipid antibodies (aPL) and anticardiolipin antibodies (aCL) were positive in 12 (32.4%) and 15 (40.5%) of the patients studied, respectively. Of these patients, five developed deep vein thrombosis (DVT), one cerebral sinus thrombosis, and one presented with purpura fulminans. In the presence of these antibodies, the risk of development of thrombosis was found to be 46.6%. The patient with purpura fulminans presented with large ecchymotic lesions on the distal parts of the extremities. Eight patients had aPL and aCL but had no thrombotic episodes.
Treatment
All six patients with thrombotic episodes in the venous system were anticoagulated with unfractionated heparin and/or low-molecular-weight heparin (LMWH). The patients with antiphospholipid syndrome (APS) and DVT were anticoagulated with LMWH for three weeks (1 mg/kg/dose, twice a day), subsequently the dosage was reduced to once a day. On the third month after the diagnosis — if residual thrombus was still present— we decided to continue anticoagulation with either LMWH or oral anticoagulation for an additional three months. At the sixth month, we assessed Doppler ultrasonographic findings, plasma D-dimer and factor VIII (FVIII) levels since residual thrombosis and high plasma D-dimer levels are important risk factors for recurrence of the thrombosis, before stopping anticoagulation.6,7 We anticoagulated six patients — five with DVT and one with cerebral sinus thrombosis— with LMWH for at least six months. No residual thrombosis was detected by Doppler ultrasonography in four with DVT on the sixth month, and anticoagulation was subsided. The anticoagulation was continued for a year due to high D-dimer, FVIII levels and strongly positive aPL antibodies in one patient. Unfortunately, DVT recurred on the other leg of this patient during oral anticoagulation therapy.
One patient with purpura fulminans was treated with plasma exchange along with immunsuppresive therapy (steroid, AZA and mycophenolate mofetil). 8 All patients with positive aPL and aCL but without thrombotic episodes were treated with aspirin.
Thrombotic microangiopathy
Thrombotic microangiopathy (TMHA) was detected in two patients. Both had low serum complement levels, positive ANA and anti-dsDNA, renal involvement with high urea and creatinine levels and hemolysis. In one of these patients, the renal biopsy showed signs of overt thrombotic microangiopathy. He had decreased renal function and had a very resistant course.
Treatment
After pulse steroid treatment, plasma exchange was performed for TMHA. In the last patient the dose of cyclophosphamide could not be given at a sufficient amount because of the low glomerular filtration rate (GFR), and rituximab was subsequently started.
Mortality
The overall mortality was 4.6% with one loss due to MAS complicated with secondary infection and one with ALL.
Discussion
Hematological manifestations of SLE have a large impact not only on the quality of life of the patient but also on survival. The spectrum of hematological findings vary in SLE. It is important to be aware of the different types of involvement since their treatment requires prompt recognition of the problem, and the treatment of each abnormality may be different.
Our treatment strategies for hematological findings of pediatric systemic lupus erythematosus patients
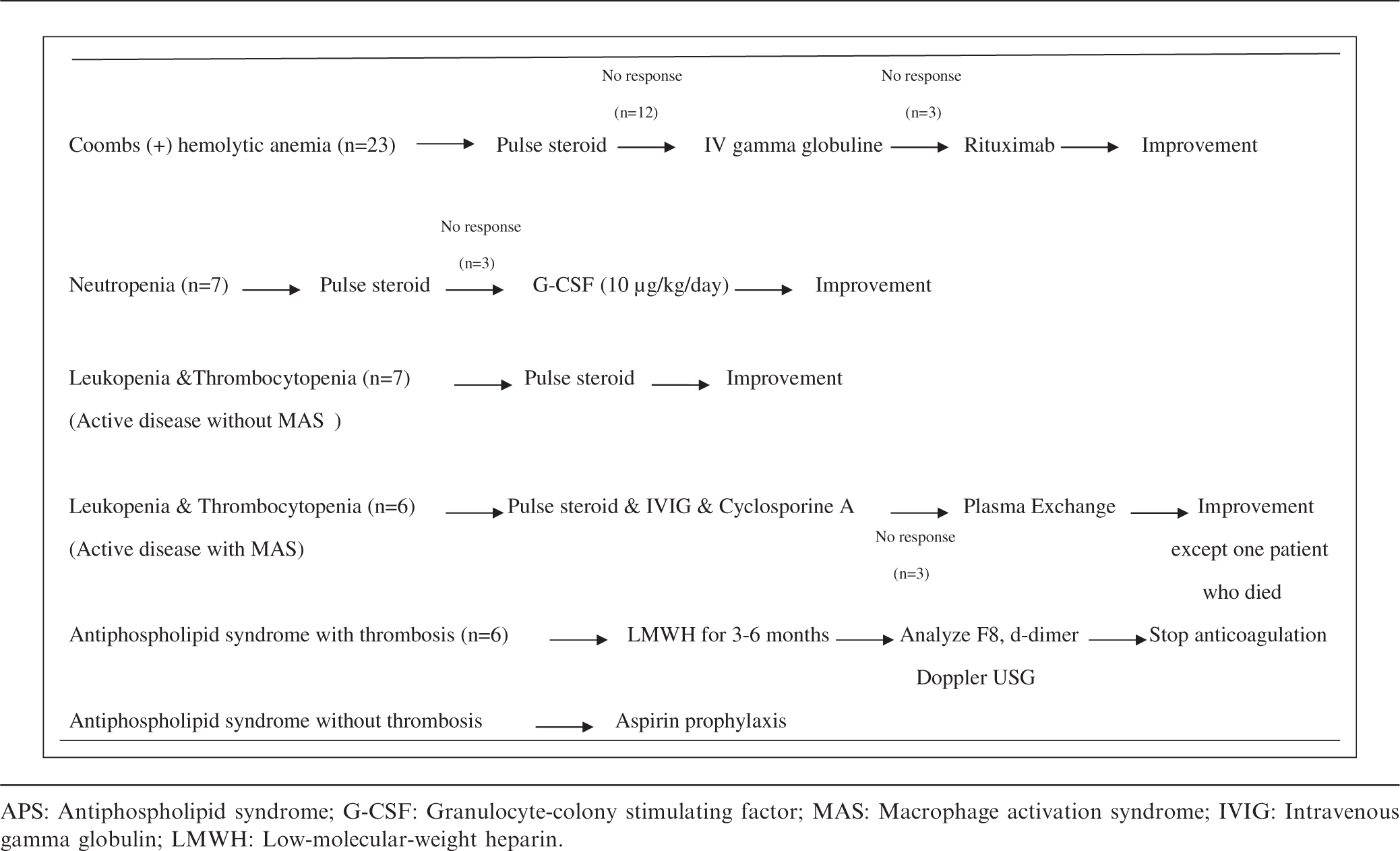
APS: Antiphospholipid syndrome; G-CSF: Granulocyte-colony stimulating factor; MAS: Macrophage activation syndrome; IVIG: Intravenous gamma globulin; LMWH: Low-molecular-weight heparin.
It should be remembered that anemia in SLE can also be due to the immunosuppressive drugs used, chronic inflammation, iron deficiency and, in cases of renal failure, erythropoietin deficiency.
In a previous retrospective study performed in 207 children with SLE aged between 2 and 16 years, leukopenia has been reported more frequently (58%) than anemia and thrombocytopenia. 17 The frequency of leukopenia was slightly lower in our cohort. The frequency of leukopenia and neutropenia were 35.1% (n = 13) and 18.9% (n = 7), respectively. Leukopenia in SLE patients is usually due to active disease and resolves as disease activity is suppressed. There is also strong evidence for the role of endogenous interferons — which increase as the disease flares up — in the pathogenesis of leukopenia. 18 If the sedimentation rate and C3 are normal along with improving clinical features, one must consider a secondary leukopenia due to the immunsuppressive drugs. Neutropenia also may develop due to anti-neutrophil antibodies, drugs, infections and MAS. In neutropenic patients, granulocyte colony stimulating factor (G-CSF) can be used to prevent infectious complications. However, flare-up of the disease has been reported with the use of G-CSF. 19 We used G-CSF in three of seven neutropenic patients after confirming the absence of underlying hematological malignancy and did not observe activation of the disease (Table 2).
Thrombocytopenia in SLE patients has a reported prevalence ranging from 7% to30%, comparable with our data. 20 Immune thrombocytopenic purpura (ITP) may precede the diagnosis of SLE in up to 16% of patients. 21 The development of the clinical and laboratory features of the disease may take 10 years in some cases. 21
The differential diagnosis of pancytopenia in SLE includes secondary hemophagocytosis or MAS. MAS is becoming increasingly recognized in SLE. 22 MAS can be very severe as was the case in one of our patients. It needs to be treated aggressively with pulse steroids and cyclosporine A, similar to MAS secondary to juvenile idiopathic arthritis (JIA). Among pediatric rheumatologic disorders, secondary HLH- MAS is most commonly seen in JIA. 23 It has also recently been reported in patients with SLE. Reported prevalence of MAS in SLE ranges from 0.9% to 4.6%. 24 Because MAS may have serious consequences and a fatal course, early diagnosis is of paramount importance. In fact in our three patients diagnosed before the development of the full clinical picture of HLH, the prognosis was excellent. In a multicenter study, it has been shown that MAS is more common in patients with SLE who present with oral/nasal ulcers, lupus nephritis, central nervous system involvement, arthritis, serositis and hematological findings. 22 Since cytopenia is not an unusual laboratory finding of SLE, it is not helpful for diagnosis of MAS as much as it is in JIA. On the other hand, fever is very distinctive for the diagnosis of MAS in juvenile SLE. On laboratory grounds, hyperferritinemia (serum ferritin > 500 ng/ml) is the strongest tool to differentiate MAS from active SLE. 22 In all of our SLE patients who developed MAS, diagnosis was established early on with the mentioned parameters. The overall MAS frequency in our group was 13.9% (n = 6), which is higher than the reported incidence in the relevant literature. Indeed the high frequency of MAS in our group may be due to increased awareness for this complication. One patient with resistant MAS died of multiorgan failure despite HLH-2004 protocol and plasma exchange. 25 Since this patient died mainly due to secondary infection, we suggest that MAS in SLE should be treated with high-dose steroids and cyclosporine plasma exchange performed as required, instead of the HLH-2004 protocol that was developed for primary hemophagocytosis.
The reported incidence of thromboembolism in pediatric onset SLE ranges from 9% to 17%. 26 The significant association between the presence of aPL and thromboembolism is well described in SLE patients. APS is termed as primary in the absence of SLE but secondary APS in its presence. Secondary APS — which is seen in approximately 30% of SLE patients— is associated with arteriovenous thrombosis, fetal loss and mild thrombocytopenia. 27 A review of 121 children showed that primary APS is usually associated with younger age of presentation and arterial thrombosis whereas secondary APS presented with venous thromboembolism in older age groups. 28 In our group, aPL and aCL were positive in 12 (32.4%) and 15 (40.5%) of the patients, respectively. A history of a thromboembolic event is probably the most significant risk factor for its recurrence in APS. In the presented patients during their follow-up, these antibodies became negative with immunsuppresive therapy in all but one. The recurrence also correlates with the titer of the antibodies and the presence of lupus anticoagulant. 29 Thrombosis recurred in only one of our patients with DVT who continued to have a positive lupus anticoagulant test whilst under anticoagulation prophylaxis. We suggest that our anticoagulation and prophlaxis regimen is an effective and safe one for SLE patients developing this complication (Table 2).
Thrombotic microangiopathy (TMHA) is a devastating feature of SLE and may be present at the onset of the disease as in one of our patients; aPL and ANA positivity, low serum C3 level, positive Coombs test and proteinuria suggested the presence of SLE. 30 All of these features were present in the two cases presented in this series. TMHA should be suspected in an ill-looking child who has purpura fulminans, thrombocytopenia and detoriation in renal function. Again, prompt treatment is important. Plasma exchange should accompany the usual immunosuppressive treatment protocol. We have also used rituximab in one child who was resistant to the initial measures. 31 The outcome of these patients has been good, although one has been diagnosed recently (two months’ follow-up).
Another important finding in the hematological spectrum encountered in SLE is dysplasia and malignant transformation. The abnormal immune system of SLE probably predisposes these patients to dysplasia. SLE is associated with an increased risk of neoplasia of the lymphoreticular system, particularly lymphoma. 32 Although Hodgkin and non-Hodgkin lymphoma are more common in these patients, leukemia has also been reported. 30 Generally, SLE precedes the hematological malignancy, but the neoplasia may develop earlier or concomitantly. In a Swedish study, doubled risk for leukemia in patients with SLE was defined. 33 Furthermore, another nested case-control study revealed that leukopenia at presentation was the foremost hematological abnormality in patients developing leukemia. 34 No association between leukemia development and renal or neurological involvement has been shown. The frequency of preceding myelodysplastic syndrome (MDS) was reported in approximately a quarter of the patients with SLE developing leukemia. 34 One of our patients unfortunately developed ALL following MDS. She also had class 3 lupus nephritis and autoimmune hepatitis, which may suggest that she had a somewhat severe form of the disease.
Several theories have been proposed to identify the association between SLE and lymphoid malignancies. A possible mechanism is the oncogene activation in the autoimmune diseases. This hypothesis has been supported by the increased expression of the proto-oncogenes c-myc and c-myb in lymphocytes of patients with SLE. 35 Immunosuppressive drugs used in the treatment may also render the SLE patients more susceptible to this complication. For example, AZA is an anti-metabolite drug that has been used in the treatment of SLE since the 1960s. It causes defective DNA mismatch repair, possibly leading to proliferation of leukemic clones. 36 In our patient, AZA was used as a part of the treatment for four months only.
This is the first study presenting an in-depth analysis of the spectrum of hematological features of pediatric-onset SLE and their treatment. Since hematological involvement constitutes one of the important elements of the clinical picture of SLE, it should be in the differential for a hematologist, especially in a child with bicytopenia/pancytopenia and thrombosis. On the other hand, since hematological involvement might be responsible for significant morbidity and mortality, pediatric rheumatology specialists should refer their pediatric patients with SLE to pediatric hematology clinics at the earliest suspicion.
Footnotes
Funding statement
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
None declared.