
Abstract
Collapsing glomerulopathy(CG), characterized by collapse of the glomerular capillary loops onto the mesangial stalks is rarely associated to systemic lupus erythematosus (SLE). Recently a genetic predisposition to HIV associated nephropathy (HIVAN) has been shown in Afro-Americans: MYH9 polymorhism in 2008 and then APOL1 variants (G1 and G2 alleles) in 2010 were shown to be strongly associated with HIVAN.
We describe here for the first time the association of CG in a young Afro-American female with SLE having a homozygous mutation of APOL1. The clinical history, laboratory findings and immunofluorescence all confirmed a diagnosis of SLE. However, studies for factors associated with collapsing glomerulopathy in other situations were consistently negative. As this Afro-American patient developed a CG, we performed genotyping of APOL1. It was found that she is homozygotic for the G2 allele of APOL1. Despite
Introduction
We report here the case of a 23-year-old Afro-American female, biopsied in France, which showed the unusual association of a collapsing glomerulopathy (CG) with lupus nephropathy. Even though various diagnostic labels have been applied, the histological features of CG have been recognized for more than 30 years. The entity designated by the name CG was initially described in large urban centres on the US East Coast in 1984 as part of the new description of HIV-associated nephropathy (HIV-AN). Initial studies 1 described in the black AIDS population a glomerulopathy considered to represent a ‘malignant’ form of focal segmental glomerulosclerosis (FSGS), clinically manifest by a severe nephrotic syndrome with rapidly progressive renal failure. Pathologically, it was characterized by collapse of the glomerular capillary loops onto the mesangial stalks, hence the name CG, with marked overlying epithelial proliferation.
The racial predilection of this disease became rapidly apparent. In Europe, the first autopsy series of white homosexuals with AIDS was negative for CG. A large series of patients with AIDS from Paris 2 revealed HIV-AN only in black African patients. By 1991, the ethnic predisposition was well established, 3 and more recently the genetic predisposition to HIV-AN has been shown in Afro-Americans: MYH9 polymorphism 4 and then APOL1 variants (G1 and G2 alleles) were shown to be strongly associated with HIV-AN. 5
Case report
This 23-year-old Afro-American female, with no familial history of renal disease, was admitted to our hospital in Paris in early April 2011 with a nephrotic syndrome. She had received a diagnosis of systemic lupus erythematosus (SLE) in 2008 with non-erosive polyarthritis, fever, lymphadenopathy and weight loss of 25 pounds over the past several weeks. She also had pancytopenia, antinuclear antibodies (ANAs) (1/1280) and anti-double-strand DNA antibodies (ds-DNA) (367.7 UI/ml). Despite proteinuria (700 mg/d) no renal biopsy was performed at that time. The patient was treated with prednisone at 0.5 mg/kg/d and hydroxyquinine 400 mg/d, with clinical and laboratory improvement. In April 2011 while on a trip to France, she presented to our emergency room with a flare of her lupus over a 3-week period with weight gain of 26 pounds, polyarthralgias, cough and dyspnoea, a papuloerythematous eruption of the face and torso and marked pedal oedema. Blood pressure was 140/90 mmHg and she was afebrile. She was pancytopenic with stigmata of haemolysis, but without schizocytes. Renal involvement was suspected because of a nephrotic syndrome (albumin 16.4 g/l, non-selective proteinuria measured at 4.6 g/d) and acute renal failure (creatinine 676 µmol/l, urea 47 mmol/l). There was no haematuria but leucocyturia was positive together with a urinary tract infection. ANA and ds-DNA were strongly positive (respectively 1/16,000 and 4000 UI/ml) and anti-phospholipid and anti-cardiolipid were negative. Renal echography revealed kidneys of normal size.
Confronted with a presumptive diagnosis of severe lupus nephritis, three 1-g boluses of methyl prednisolone were given, followed by oral prednisone at 1 mg/kg/d. She was dialysed on admission and 2 days later developed an epileptic crisis necessitating transfer to intensive care. Anti-epileptic treatment with continuation of the corticotherapy and dialysis sessions was efficacious. A cerebral MRI performed a week later showed the presence of a semi-recent infarct in the left internal parietal region, interpreted as corresponding to cerebral lupus vasculitis. A transjugular renal biopsy was performed 12 days after admission.
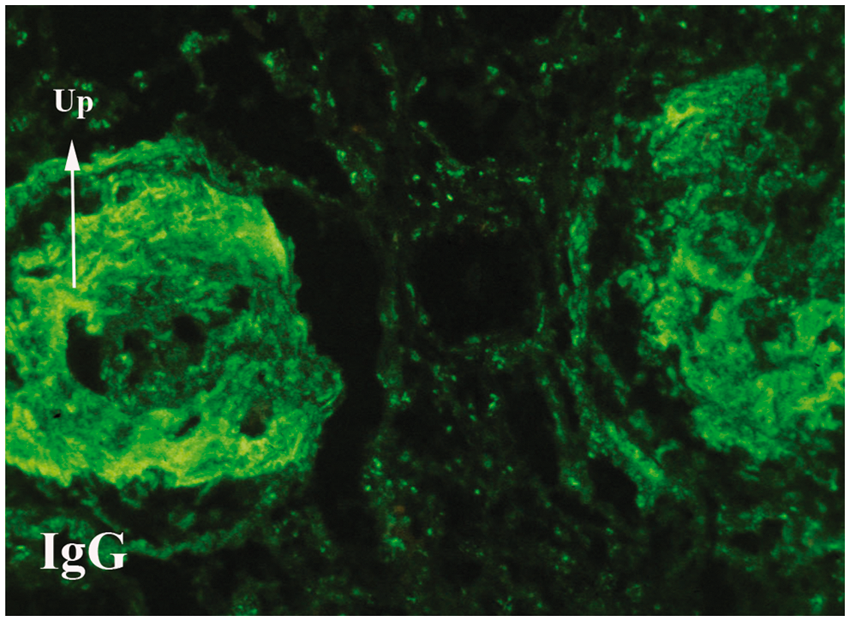
The renal biopsy contained 15 glomeruli, two of which were obsolescent. Most glomeruli displayed segmental or global collapse with sclerosis. Two of the remaining glomeruli had a crown of tall, markedly swollen and vacuolated epithelial cells, some containing hyaline droplets, covering the collapsed tuft (Figure 1). Numerous vacuolated cells of presumptive epithelial origin lay free in Bowman’s space. There were no segmental or diffuse proliferative glomerular lesions, nor endomembranous or extramembranous immune deposits visible on light microscopy. There was widespread tubular atrophy, but without microcytic change, and interstitial fibrosis with only modest interstitial inflammation. Vessels were normal. Direct immunofluorescence showed strong granular staining of glomeruli and in a vascular and tubulointerstitial pattern with predominant staining with polyclonal anti IgG, anti-C1q and anti-C3 (Figure 2). There was also prominent nuclear staining, typical of lupus nephritis. Because of limited material in this transjugular biopsy, it was not possible to perform immunohistochemical staining to characterize the vacuolated cells overlying the collapsed tufts. The final impression was collapsing glomerulopathy superimposed on glomeruli having immunofluorescence consistent with SLE, although without proliferative lesions or necroses by light microscopy.
Collapsing glomerulopathy. (A) Glomerulus with segmental sclerosis and collapse on left, but no significant proliferative lesions, covered over by a halo of tall epithelial cells, some vacuolated. Similar cells float free in Bowman’s space. (B) Second glomerulus at high magnification showing glomerular tuft overlain by tall, basophilic epithelial cells frequently containing large vacuoles. Masson trichrome stain, A: × 450; B: × 650. Immunofluorescence for IgG. Two glomeruli with advanced sclerotic lesions showing diffuse staining for IgG, seen to be granular in the right glomerulus with less advanced lesions. Speckled staining of nuclei in tubulointerstitium. x200.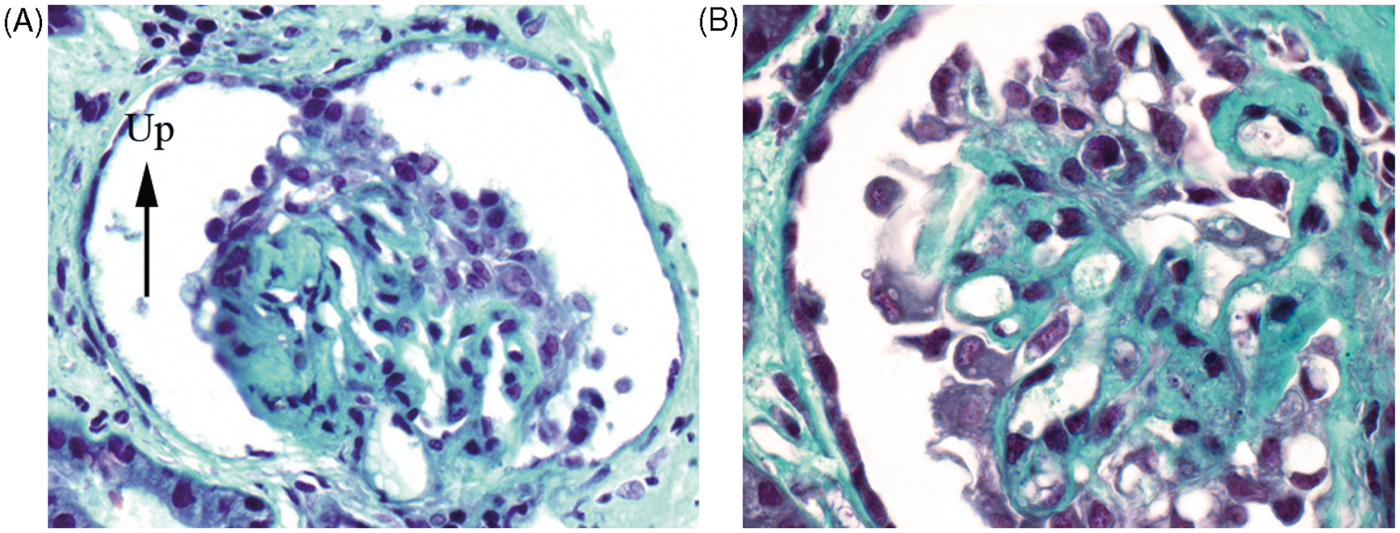
The clinical history, laboratory findings and immunofluorescence all confirmed a diagnosis of SLE. However, studies for factors associated with collapsing glomerulopathy in other situations (HIV, cytomegalovirus, parvovirus B19, hepatitis C virus, drug abuse and pamidronate medication) were consistently negative. As this Afro-American patient developed a CG, we performed genotyping of APOL1. It was found that she is homozygotic for the G2 allele of APOL1.
Her subsequent course was complicated days later by haemoptysis, and diffuse alveolar haemorrhage was confirmed by bronchioalveolar lavage. Immunosuppressive treatment associating cyclophosphamide, 500 mg every other week, was started as well as three plasma exchanges. On this regimen her neurological and haematological symptomatology improved, but she remains on dialysis 6 months after her initial presentation.
Discussion
We report here an association of CG related with APOL1 polymorphism in an Afro-American lupus patient. The classification of CG comprises three groups: idiopathic, genetic, and secondary or reactive, according to the terminology. 6 The association of CG and SLE is rare, eight cases having been reported in the literature. A recent study described 19 patients with lupus-associated CG. This largest series was composed of a majority of black women. 7 Indeed, in this study, the authors evoked the possibility of the role of APOL1 allelic variants in these patients. CG has a known predilection for blacks in other situations and this seems to be the case with lupus as well. Allelic variants of APOL1 have been associated with HIV-AN in Afro-American patients, as well as with other forms of FSGS. 5
To the best of our knowledge, our patient is the first in whom the presence of homozygosity for the G2 allele of APOL1 has been demonstrated in the context of a CG not due to HIV-AN. Similarly, two Afro-American patients have been described who had a CG developing in the context of a C1q nephropathy, associated with E1 haplotype of MYH9. 8 MYH9 and APOL1 are very closely linked at the same locus on chromosome 22 and both are closely correlated with the occurrence of HIV-AN. Independently of its immediate aetiology, the CG pattern of podocytopathy seems tied to the allelic variants of APOL1 in black patients. It seems likely that in a person carrying these alleles, a variety of renal triggering factors, such as HIV, SLE or C1 nephropathy are capable of inciting CG.
Regarding treatment, CG has historically been resistant to standard therapies for FSGS, with rapid evolution to end-stage renal failure. Follow-up from 13 patients from the Salvatore et al. study revealed that seven patients progressed to ESRD at the time of biopsy up to 21 months later, one patient had complete remission and five patients displayed partial remission. 7 Thus, the general impression in the literature is one of a more favourable course of lupus-associated CG than for idiopathic CG. Our patient did not respond to the immunosuppressive treatment and remains on chronic haemodialysis therapy. Perhaps the question is one of timing. In HIV-AN, early institution of antiretroviral therapy may lead to complete remission of the nephrotic syndrome, 9 improvement in the biopsy lesions of HIV-AN and slow the progression to ESRD. 10 The question arises as to whether therapy directed toward the lupus would have the same effects on lupus-associated CG.
Our Afro-American patient carries a homozygous mutation of APOL1, whose association with HIV-AN has previously been shown. She developed CG in the context of a flare of lupus. This is a rare complication, but suggests that this pattern of podocyte injury is related to aggression of the podocytes carrying this homozygous mutation. Rapid treatment of the SLE may be essential to breaking this cycle. Future studies will be necessary to fully understand the role of APOL1 in the pathology of collapsing glomerulopathy.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
The authors have no conflicts of interest to declare.