
Abstract
Objective
Studies in animal models have indicated that Pellino 1 is involved in inflammatory and autoimmune diseases, such as systemic lupus erythematosus (SLE). The current study was designed to determine whether PELI1 confers genetic susceptibility to SLE in humans, as assessed in a Chinese Han population.
Methods
Blood samples were drawn from patients diagnosed with SLE and healthy volunteers. Three single nucleotide polymorphism (SNP) loci with a minor allele frequency of at least 0.05 were chosen to evaluate the correlation between PELI1 genotype and the incidence of SLE.
Results
There was a significant difference in the frequency distribution of the rs329497 allele between the SLE patients and the healthy controls (A vs. G; Bonferroni corrected p = 0.036, odds ratio = 0.75, 95% confidence interval = 0.60–0.94). No differences in the genotype and allele frequencies of other SNP loci were observed between the two groups. Furthermore, the alleles and genotypes of the three SNPs were not associated with lupus nephritis.
Conclusion
In the Chinese Han population, PELI1 SNPs may be associated with SLE susceptibility.
Introduction
Systemic lupus erythematosus (SLE) is a chronic inflammatory autoimmune disease characterized by damage to multiple organ systems and production of autoantibodies. SLE typically presents as a skin and joint disorder in women between 20 and 45 years of age, observed as a butterfly rash on the face, with low-grade fever and non-deforming arthritis. The most common severe outcome is nephritis, but other tissue and organs systems can be affected. There is a higher incidence of SLE among Asian compared to Caucasian females, with an incidence of approximately 100 in 100,000. 1
It is widely acknowledged that the etiology of SLE is multifactorial in nature, and recent genome-wide association studies (GWASs) suggest that genetic predisposition plays a critical role.2,3 The complex pattern of inheritance in SLE indicates that multiple genes contribute, 4 particularly those within the human leukocyte antigen locus. Genes within other loci involved in the regulation of the immune response may also contribute to susceptibility for and development of SLE, including integrin alpha M, protein tyrosine phosphatase, non-receptor type 22, and interferon-regulatory factor 5.5,6
A recent GWAS in Korea reported that the 2p13.3 region encompassing PELI1 is a susceptibility locus for Kawasaki disease, which is characterized an abnormal immunologic reaction to an infection.7,8 PELI1 is located on chromosome 2p13.3, producing an mRNA of 3780 bp, which is ubiquitously expressed in most organs and is involved in the pathogenesis of inflammatory and autoimmune diseases.7,9–11 The gene encodes the protein Pellino 1, a member of the RING finger E3 ubiquitin ligase family, which is a key component in many signal transduction pathways, and can be phosphorylated by interleukin receptor-associated kinases (IRAK) 1 and 4, or IκB kinase ɛ/TANK-binding kinase 112,13 Pellino 1 function is thought to involve the interleukin-1 and Toll-like receptor (TLR)-signaling pathway, specifically through interactions with the tumor necrosis factor receptor-associated factor 6 and IRAK proteins. Reports have also associated Pellino 1 with responses to lipopolysaccharide and dsDNA, regulation of B-cell and cytokine production, and the TLR4 pathway.
Chang et al. reported that Peli1-deficient mice spontaneously developed lupus characterized by renal damage and autoantibody production via dysregulation of c-Rel, 11 a member of the NF-κB family of transcription factors. Furthermore, Peli1 is required for TLR3-stimulated expression of pro-inflammatory cytokines and B-cell activation. 10 Peli1 also has a critical role in other TLR signaling pathways and regulates activation of NF-κB through an interaction with IRAK1, which is also a potential susceptibility gene for the development of SLE.14,15 NF-κB and IRAK1 have been implicated in the pathogenesis of SLE in mice and humans.16–20
To our knowledge, no previous research studies have reported PELI1 polymorphisms in SLE pathogenesis. As Pellino 1 expression is critical for many immune regulation pathways, we hypothesized that PELI1 may be involved in the development of SLE. To examine this, we compared three single nucleotide polymorphisms (SNPs) (rs329497, rs329498, and rs10496105) and their association with SLE susceptibility.
Materials and methods
Patients and sample collection
This case-control study examines the association between PELI1 polymorphisms and SLE susceptibility. Patients with SLE (n = 395; 10 men and 385 women, mean age 33.1 ± 11.6 y) were recruited from the Southwest Hospital of the Third Military Medical University in Chongqing and the Affiliated Hospital of Guilin Medical College. All the patients fulfilled at least four of the American College of Rheumatology 1997 Revised Criteria for the classification of SLE. 21
For comparison, healthy individuals (n = 378; 19 men and 359 women; mean age 34.0 ± 7.6 y) were included as controls. Blood samples were collected from the controls during regular health examinations at the Medical Examination Center of Southwest Hospital. None of the healthy individuals had abnormal clinical examinations, and all were free of clinical SLE symptoms and history of rheumatism or immune disease. All SLE patients and the control group participants were Chinese Hans.
Phenotypic characteristics of SLE patients, n = 323

Abbreviations: dsDNA, double stranded DNA; SSA, Sjogren’s syndrome-A; SSB, Sjogren’s syndrome-B; Sm, Smith; U1RNP, U1 ribonucleoprotein.
SNPs selection criteria
The full-length human PELI1 data for the Chinese Han population of Beijing, China, was downloaded from the international HapMap project databank. All PELI1 tag-SNPs with a minor allele frequency (MAF) ≥ 0.05 and linkage disequilibrium (r2 > 0.8) in the Chinese Han population were determined using the Haploview 4.2 haplotype analysis software (Daly Lab, Cambridge, MA, USA). Finally, the tag-SNPs information was obtained from the National Center for Biotechnology Information (NCBI) databank (US National Library of Medicine, Bethesda, MD, USA). SNPs located within the coding portion of the gene, including both synonymous and non-synonymous substitutions, were also used to identify PELI1 SNPs.
The tag-SNPs offer great improvements in efficiency and information capture of a large fraction of all variations that may exist; use of SNPs with MAF <5% limits the power of current statistical tests of association.22,23 It is well known that SNPs in the coding region are more likely to lead to functional effects. Non-synonymous SNPs can potentially affect the function of the protein, subsequently altering the carrier’s phenotype by disrupting interaction with other proteins.24,25 Synonymous (s)SNPs, on the other hand, do not affect the amino acid sequence of the encoded protein but may still have significant effects on a protein’s functional capacity;
26
for example, sSNPs may affect mRNA splicing, mRNA folding, stability and regulation of translation.27,28 Thus, we selected three tag-SNPs located in the exonic sequences of PELI1 (sSNPs: rs329497, rs329498, and rs10496105) (Figure 1).
Graphical overview of the PELI1 genomic region and positions for single nucleotide polymorphisms. White boxes, exons; gray boxes, 5′ untranslated region (UTR) and 3′ UTR; black lines, introns; arrow, direction of transcription. PELI1 has seven exons; rs329497 and rs329498 are both located in the fourth exon, and rs10496105 is located in the seventh exon.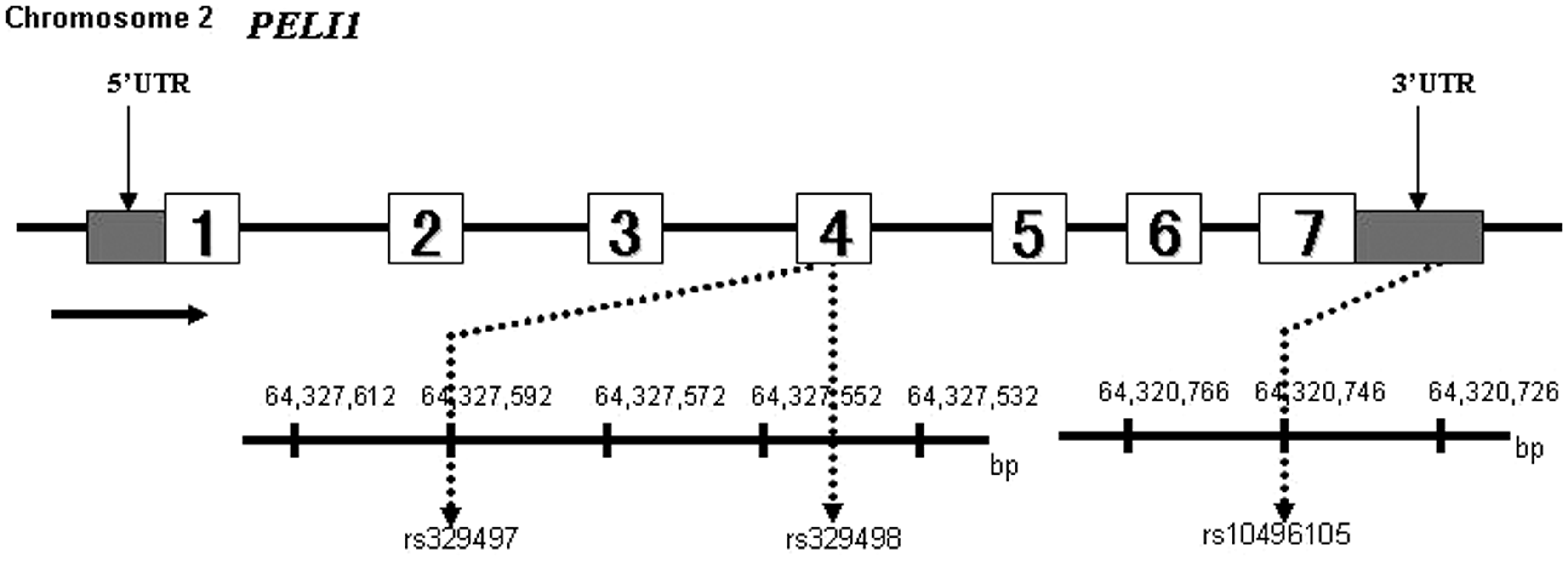
DNA samples and genotyping assays
Peripheral blood (2 mL) was collected from each participant with EDTA anti-coagulation and stored at −80℃ until use. Genomic DNA was then extracted using a Sangon genomic DNA extraction kit (Sangon Biotech, Shanghai, China) according to the manufacturer’s instructions. The amplification primers were as follows (forward and reverse): rs329497: 5′-ACGTTGGATGGATGTTGTAATACCTGAAA-3′ and 5′-ACGTTGGATGGACCAGCATAGCATATCATA-3′; rs329498: 5′-ACGTTGGATGGATGTTGTAATACCTGAAA-3′ and 5′-ACGTTGGATGGACCAGCATAGCATATCATA-3′; rs10496105: 5′-ACGTTGGATGTCCTTAAGTCTCACTGTGGC-3′ and 5′-ACGTTGGATGATAGTGCCCCCGTTTTTAGC-3′. The single-strand extension primers were as follows: rs329497: 5′-CCCACCCACCACAGTCTGGGCCCGAGA-3′; rs329498: 5′-ATGTTGTAATACCTGAAACATATC-3′; rs10496105: 5′-GACTGTGGCAATCTCCTTA-3′.
For genotyping of the three SNP loci, SNP sequence-specific extension primers were added into the PCR amplification products (Sangon Biotech). Briefly, 1 µL of the DNA sample (at 5 ng/µL) was combined with 0.95 µL of water, 0.625 µL of PCR buffer containing 15 mM MgCl2, 1 µL of 2.5 mM dNTP, 0.325 µL of 25 mM MgCl2, 1 µL of PCR primers, and 0.1 µL of 5 units/µL HotStartTaq (Qiagen, Venlo, Netherlands). The reaction was incubated at 94℃ for 15 min followed by 45 cycles at 94℃ for 20 s, 56℃ for 30 s, and 72℃ for 1 min, and a final incubation at 72℃ for 3 min. After PCR amplification, the remaining dNTPs were dephosphorylated by adding 1.53 µL of water, 0.17 µL of SAP buffer, and 0.3 U of shrimp alkaline phosphatase (Sequenom, San Diego, CA, USA). The reaction was placed at 37℃ for 40 min, and the enzyme was deactivated by incubating at 85℃ for 5 min. The single primer extension over the SNP was then combined with 0.755 µL of water, 0.2 µL of 10 × iPLEX buffer, 0.2 µL of termination mix, 0.041 µL of iPLEX enzyme (Sequenom), and 0.804 µL of 10 µM extension primer. The single base extension reaction was carried out at 94℃ for 30 s and then 94℃ for 5 s, followed by 5 cycles of 52℃ for 5 s and 80℃ for 5 s, total 40 cycles, then 72℃ for 3 min. The reaction mix was desalted by adding 6 mg of cation exchange resin (Sequenom), mixed and re-suspended in 25 µL of water. The completed genotyping reactions were spotted onto a 384-well SpectroCHIP (Sequenom) using the MassARRAY Nanodispenser (Sequenom) and determined by matrix-assisted laser desorption ionization time-of-flight mass spectrometry. Genotype determination was performed in real time with MassARRAY RT software version 3.0.0.4 and analyzed using the MassARRAY Typer software version 3.4 (Sequenom). The genotyping success rate was >95%.
Statistical analysis
Data were analyzed with SAS 9.2 software for Windows (SAS Institute Inc., Cary, NC, USA). The chi-square (χ 2 ) test was used to determine the extent of deviation for genotype frequencies of each SNP in the controls from those expected under Hardy–Weinberg equilibrium (HWE). Allele, genotype and haplotype frequencies were compared between the SLE patients and healthy individuals using the χ 2 test and logistic regression model, and the p values were corrected (pc) with the Bonferroni correction by multiplying the number of analyses performed. The SLE group was further stratified by clinical evidence of nephritis. All p values were three-tailed, and a p < 0.05 was regarded as significant.
The sub-haplotype (subsets of alleles from the full haplotype) analyses were carried out using the Haplo.stats package for R (Cran.r-project.org). To evaluate the association of sub-haplotypes with SLE risk, a sliding window of two-loci alleles was evaluated across the entire haplotype using the “haplo.score.slide” function, and the “haplo.cc” function was used to calculate the frequency of sub-haplotype between cases and controls and odds ratios (ORs).29,30
Results
Genotype and allele frequency distribution of PELI1 polymorphisms
Distribution of genotype and allele frequencies and association analysis of PELI1 polymorphisms in cases and controls
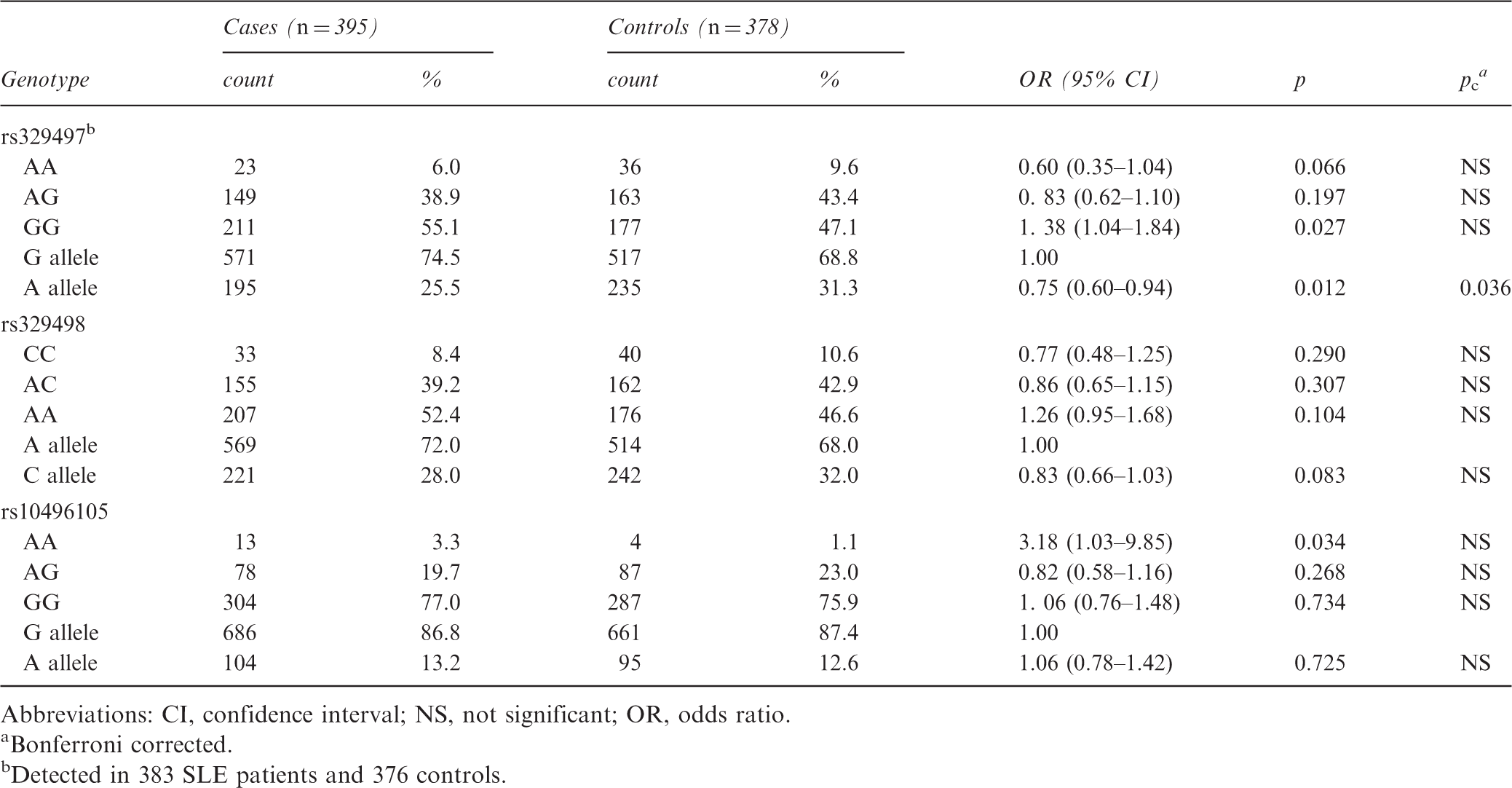
Abbreviations: CI, confidence interval; NS, not significant; OR, odds ratio.
Bonferroni corrected.
Detected in 383 SLE patients and 376 controls.
Haplotype analysis
The three SNPs analyzed in this study were under linkage disequilibrium (Figure 2). The haplotypes existed in the sequence of rs329497–rs329498–rs10496105. The frequencies of different haplotypes and the results from association analysis with SLE are presented in Supplementary Table 1. All haplotypes that had frequencies of >5 were chosen for analysis in order to simplify the statistical comparisons. G-A-G, the most common haplotype found in the healthy controls, was set as the reference for association analysis of specific haplotypes with SLE. The frequency of the haplotype A-C-G in SLE patients was significantly lower than that in the healthy controls (A-C-G vs. G-A-G, p = 0.001; OR = 0.63, 95% confidence interval (CI) = 0.48–0.84) (Supplementary Table 1). Of the three tested SNPs, only the A allele of rs329497 was associated with SLE (Table 2); thus, conditioning the logistic regression model for the associated SNP rs329497 necessitated that rs329497 and all constructed haplotypes be included as independent variables in order to accurately test whether, and to what extent, the haplotypic association may be affected by rs329497 (via the Genetic Module of SAS software). Testing of the model showed that the significant difference remained for the frequency of the A-C-G haplotype (pc = 0.001).
Haplotype block structure of the PELI1 single nucleotide polymorphisms with consistent associations among racial groups in the study cohort. The linkage disequilibrium block structure was analyzed using Haploview software, version 4.2. The r2 color scheme is as follows: white (r2 = 0), gray (0 < r2 < 1), black (r2 = 1).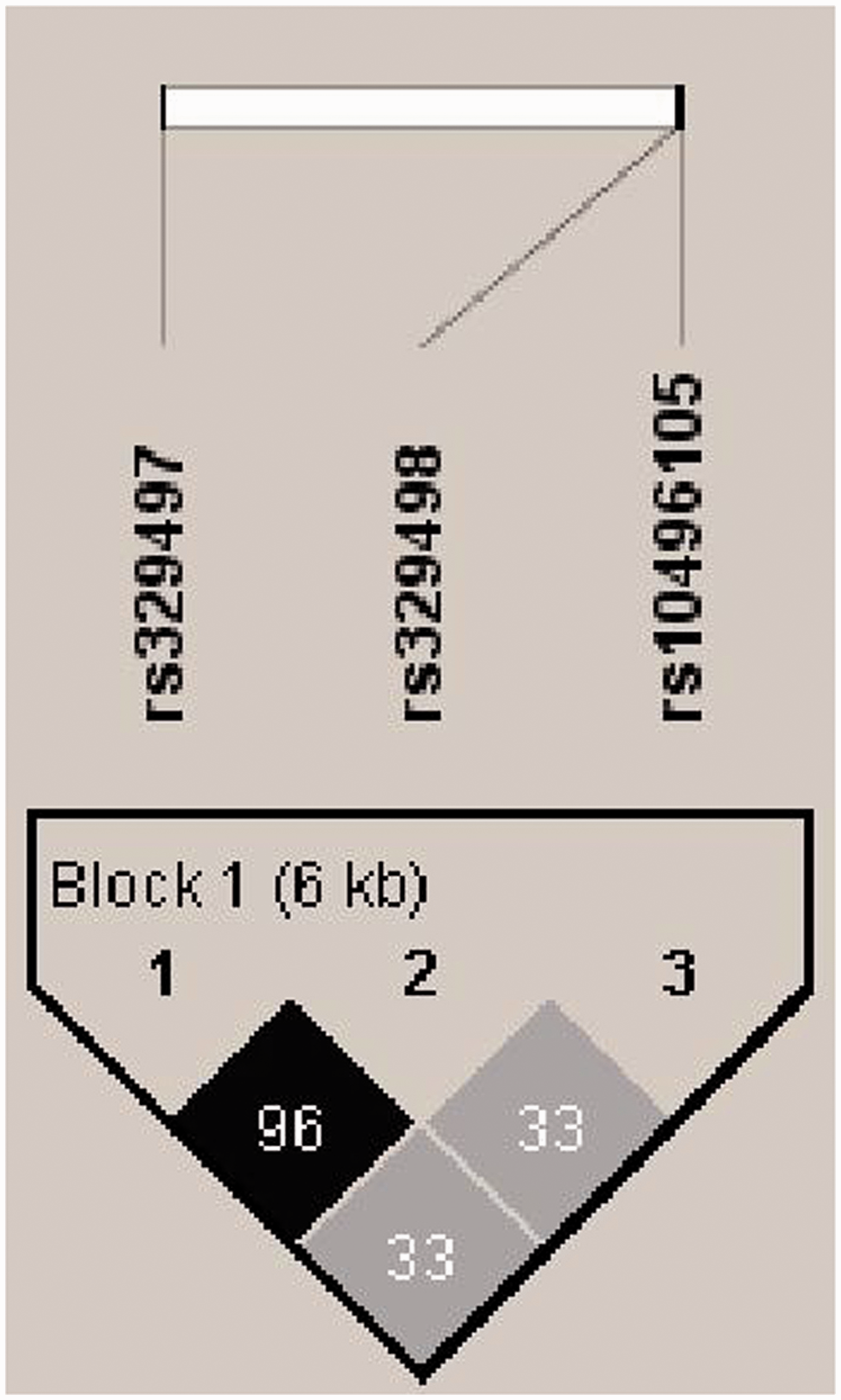
To confirm the effect of rs329497, the haplo.score.slide was used to evaluate the association of sub-haplotypes with SLE risk. The results are shown for groups of the three SNPs in Supplementary Figure 1. The significant sub-haplotype contained both rs329497 and rs329498; however, the other sub-haplotype that contained rs329498 and rs10496105 showed no significant difference between the cases and controls. Sub-haplotype analysis of rs329497 and rs329498 showed that individuals with the A-C haplotype had a significantly lower risk for SLE, compared with the common haplotype G-A (p = 0.007; OR = 0.75, 95% CI = 0.60–0.94). Thus, while the haplotypes appear to involve all three SNPs, the haplotypic association itself appears to be mainly driven by rs329497. Finally, we also found no significant differences between the cases and controls for the other haplotypes.
Lupus nephritis stratification
We then considered the possibility PELI1 SNPs would contribute to more severe disease among the SLE patients. Consequently, we examined the distribution of PELI1 genotypes stratified by lupus nephritis (Supplementary Table 2). Our data showed that no differences existed in the genotype and allele frequencies of the three loci in PELI1 between the two cohorts (p > 0.05).
Discussion
This study identifies a difference in the allele frequencies of rs329497 between the SLE patients and a control population, suggesting that the A allele may be a protective factor for SLE. This SNP is a synonymous mutation in the fourth exon. Recent findings suggest that synonymous substitutions may substantially contribute to the risk for some human diseases and other complex traits. They may bring about changes in the phenotype by affecting splicing accuracy, translation fidelity, mRNA structure and protein folding.26–28 Additional future studies will verify whether genetic variations of PELI1 influence the expression and function of Pellino 1, and will further clarify the mechanism of PELI1 in the pathogenesis of SLE.
Haplotypes are often used by investigators to pinpoint a disease-causing locus, marking the sites of recombination events in both family-based and population-based studies. In our study of SLE haplotypes, the SNPs were in the sequence rs329497–rs329498–rs10496105 (Supplementary Table 1). Our finding of the significant difference that exists among the frequency of the A-C-G haplotype in particular suggests the possibly of its acting as a protective factor against SLE; however, logistic regression model analysis of all three SNPs and sub-haplotype analyses revealed that the haplotypic association is largely dependent upon rs329497 (Supplementary Table 1, Supplementary Figure 1
A search of the National Human Genome Research Institute and NCBI databases found no reports of the three loci in any of the 14 SLE GWASs published to date.5,31–45 Some of these studies genotyped more than 300,000 tag-SNPs using the Illumina platform, thus their efforts were expected to have included the three SNPs included in our study. However, GWAS demand higher significance levels, with the p threshold being set at 10−5 or below, thus the three SNPs may not have been associated with SLE in those studies because they failed to meet the this threshold. SLE pathogenesis is believed to involve the combined effects of a large number of minor genes. While each allele gene may only make a minor contribution (OR ∼1.5), the accumulative effect of several genes may then increase the risk of SLE. 46 Thus, researchers should pay attention to these minor genes in their efforts to elucidate the pathogenic mechanisms of SLE and develop effective targeted therapies or preventive measures. Although the sample size was small compared with the previous GWAS, our study provides novel findings as it was specifically focused on the association of PELI1 with SLE and we performed a stratification analysis for lupus nephritis.
Our study has several limitations that should be noted. First, our results found only one SNP (rs329497) with allele frequencies that can be associated with SLE. Second, the association analyses between the three SNPs and lupus nephritis produced negative results, possible due to the small sample size and reduced statistical power in which to detect a true association. Multiple replications in large samples would provide the most straightforward path to identifying robust and broadly relevant associations. 23 Patient populations of African, Caucasian, Japanese, and Chinese descent contain varying histories of mutation, migration, isolation, and genetic drift, and there is no doubt that there should be substantial differences in allele frequencies among these different ethnic groups. 47 Thus, our results should not be generalized to other populations. Different ethnic groups should be examined to determine if PELI1 polymorphism results in associations with autoimmune disease.
As we know, autoimmune diseases, such as rheumatoid arthritis and SLE, are considered chronic inflammatory diseases caused by abnormally activated T cells and dysregulation of TLR pathways.48,49 Recent studies have indicated that Pellino 1 is a member of the RING finger E3 family of proteins with important roles in the regulation of both innate immune receptor signaling and T-cell tolerance. 50 The in vivo T-cell activation seen in Peli1-deficient mice is very likely due to the loss of peripheral T-cell tolerance. Peli1 deficiency may also have other contributions to autoimmunity, including elevated antinuclear autoantibodies and immunoglobulin deposition in kidney glomeruli. 11 A recent study by Enesa et al. identified Peli1 as a novel component of the signal transduction network by which the TLR pathway induced interferon production. 51 Xiao et al. found that Peli1 was crucial for the pathogenesis of multiple sclerosis in the animal model of experimental autoimmune encephalomyelitis. 9 Taken together, these data suggest that PELI1 plays a role in the pathogenesis of SLE, possibly by regulating immune signaling pathways and the function or activity of some immune cell subsets.
SLE, as an autoimmune disease, has a high level of genetic heterogeneity, and it is important to note that our results represent the Chinese Han population. Furthermore, PELI1 is a member of a constellation of genetic factors that may contribute to the pathogenesis of SLE. Nonetheless, our study provides new clues towards further elucidating the pathogenic processes of SLE, and may provide insights into the disease process in various ethnicities.
Footnotes
Funding
This work was supported by the National Natural Science Foundation of China (grant number 81271753).
Conflict of interest statement
The authors have no conflicts of interest to declare.