
Abstract
Objectives
The objective of this study was to investigate risk factors of microvascular involvement and survival in Chinese patients with primary antiphospholipid syndrome.
Methods
In this single-center, retrospective study, we enrolled 112 patients with a confirmed diagnosis of primary antiphospholipid syndrome who were admitted to Peking Union Medical College Hospital from January 2004 to December 2016. Demographic data, clinical characteristics, laboratory results, and follow-up records were collected.
Results
A total of 112 patients with primary antiphospholipid syndrome were studied. Microvascular involvement was identified in 21 patients (18.75%). Patients with microvascular involvement experienced fewer episodes of arterial or venous thrombosis (28.6% vs. 84.6%) and a higher incidence of thrombocytopenia (85.7% vs. 54.9%), respectively. Low complement and elevated high-sensitivity CRP levels were observed more frequently in the microvascular group compared with the non-microvascular group (complement 38.1% vs. 18.7%; high-sensitivity CRP 71.4% vs. 31.9%, respectively). Anti-β2-glycoprotein I antibodies were more prevalent in patients with microvascular involvement than in patients without (66.7% vs. 33.0%, respectively). Multivariate logistic regression analysis revealed that thrombocytopenia (odds ratio = 4.523, 95% confidence interval 1.139–17.962), elevated high-sensitivity CRP levels (odds ratio = 6.385, 95% confidence interval 1.969–20.704), and anti-β2-glycoprotein I antibody positivity (odds ratio = 5.042, 95% confidence interval 1.555–16.352) were independent risk factors for microvascular involvement. A Kaplan–Meier analysis revealed that survival was significantly poorer in patients with microvascular involvement compared with patients without (p = 0.0278).
Conclusions
In addition to arterial and venous thrombosis, antiphospholipid syndrome can affect the microvasculature of select organs. It is thus important for clinicians to be aware that antiphospholipid syndrome-associated microvascular involvement has a unique pathogenesis and can be a life-threatening condition.
Introduction
Antiphospholipid syndrome (APS) is an acquired autoimmune thrombophilia characterized by recurrent vascular thrombosis and/or pregnancy morbidity in the persistent presence of moderate to high titers of antiphospholipid antibodies. In addition to veins and arteries, micro-vessels may also be involved in APS. In several severe complications of APS, including hemolysis, elevated liver enzymes, and low platelet count (HELLP) syndrome, thrombotic thrombocytopenia purpura, hemolytic uremic syndrome, catastrophic APS (CAPS), and disseminated intravascular coagulation, the prevalence of microvascular abnormalities are referred to as microangiopathic antiphospholipid-associated syndromes (MAPS).1,2 These clinical manifestations depend on the type and extent of vasculature affected and the organs involved. Extensive vascular occlusion or vital organ involvement may worsen the prognosis of APS.
Microvascular involvement has been reported in patients with antiphospholipid antibodies and/or APS, but its pathogenesis has not been fully elucidated. 3 Yet very little research has compared MAPS with classical APS. Only a few case reports of APS with microangiopathy are available in the literature.4–6 The aim of this cohort study was to compare the clinical manifestations, laboratory features, and outcomes of primary APS patients with and without microvascular involvement.
Methods
Data collection
Patients with primary APS who were consecutively hospitalized in Peking Union Medical College Hospital from January 2004 to December 2016 were enrolled in our study. Patients who had incomplete medical records were excluded. A diagnosis of APS was based on the revised Sapporo APS Classification Criteria (also called the Sydney criteria). 7 A diagnosis of CAPS was based on the evidence of involvement of three or more organs in a short period of time with circulating aPL. 8 Microvascular involvement (also called MAPS) 9 was diagnosed either by histologic evidence of small vessel occlusions (such as kidney biopsy), or the presence of thrombocytopenia and microangiopathic hemolytic anemia (hemoglobin <12 g/dl, the presence of ≥2/highest possible frequency schistocytes on peripheral blood smear, lactate dehydrogenase levels ≥1.5 times the upper limit of normal range), without other underlying causes including inherited thrombotic thrombocytopenic purpura (TTP), Shiga-toxin-induced hemolytic uremic syndrome, severe infection or drug-induced thrombotic microangiopathy (TMA), or conditions such as hemolytic anemia or thrombocytopenia that were caused by primary APS (PAPS) itself. Antiphospholipid antibodies were evaluated in all patients during their first visit and rechecked at least 12 weeks later. Thrombotic events were confirmed by contrast computed tomography, ventilation-perfusion (quotient) ratio, magnetic resonance angiography, and vascular ultrasonography. Exclusion criteria included a history of connective tissue disease, such as systemic lupus erythematosus, Sjögren syndrome, and rheumatoid arthritis, systemic vasculitis, or malignancy.
Anticardiolipin antibodies (aCL) (IgG or IgM isotype) and anti-β2-glycoprotein I (anti-ß2GPI) antibodies (IgG or IgM isotype) were tested by enzyme-linked immunosorbent assay. Medium to high titers (40 units for aCL or anti-ß2GPI) were considered positive. Lupus anticoagulant (LA) activity was detected by coagulation assays according to the guidelines of the International Society on Thrombosis and Hemostasis, using the dilute Russell's viper venom time during the screening process.
Statistical analyses
Results are expressed as the mean ± standard deviation (parametric data) or median values and ranges (nonparametric data). Student's t-tests were used to compare continuous variables, whereas chi-squared tests were used to compare categorical data. Factors related to microvascular involvement were identified by multivariate logistic regression analyses (forward stepwise likelihood ratios). The cumulative probability of survival was assessed using the Kaplan–Meier method. For patients who died or were lost to follow-up, data on survival were censored at the time of death and last clinic visit or hospitalization, respectively. We compared the cumulative survival rate of patients with and without microvascular involvement by the log-rank test. A p value less than 0.05 was considered statistically significant. Statistical analysis was carried out using the SPSS statistical software package, version 22.0 (SPSS Inc., Chicago, IL, USA) and Prism, version 7.01 (GraphPad Software, San Diego, CA, USA).
Results
We reviewed a total of 801 patients hospitalized in our center with APS from January 2004 to December 2016. Primary APS was present in 260 (32.5%) cases. Only 112 PAPS with complete medical records were included in the final analysis.
Baseline characteristics and prevalence of thrombosis
Demographic and clinical data
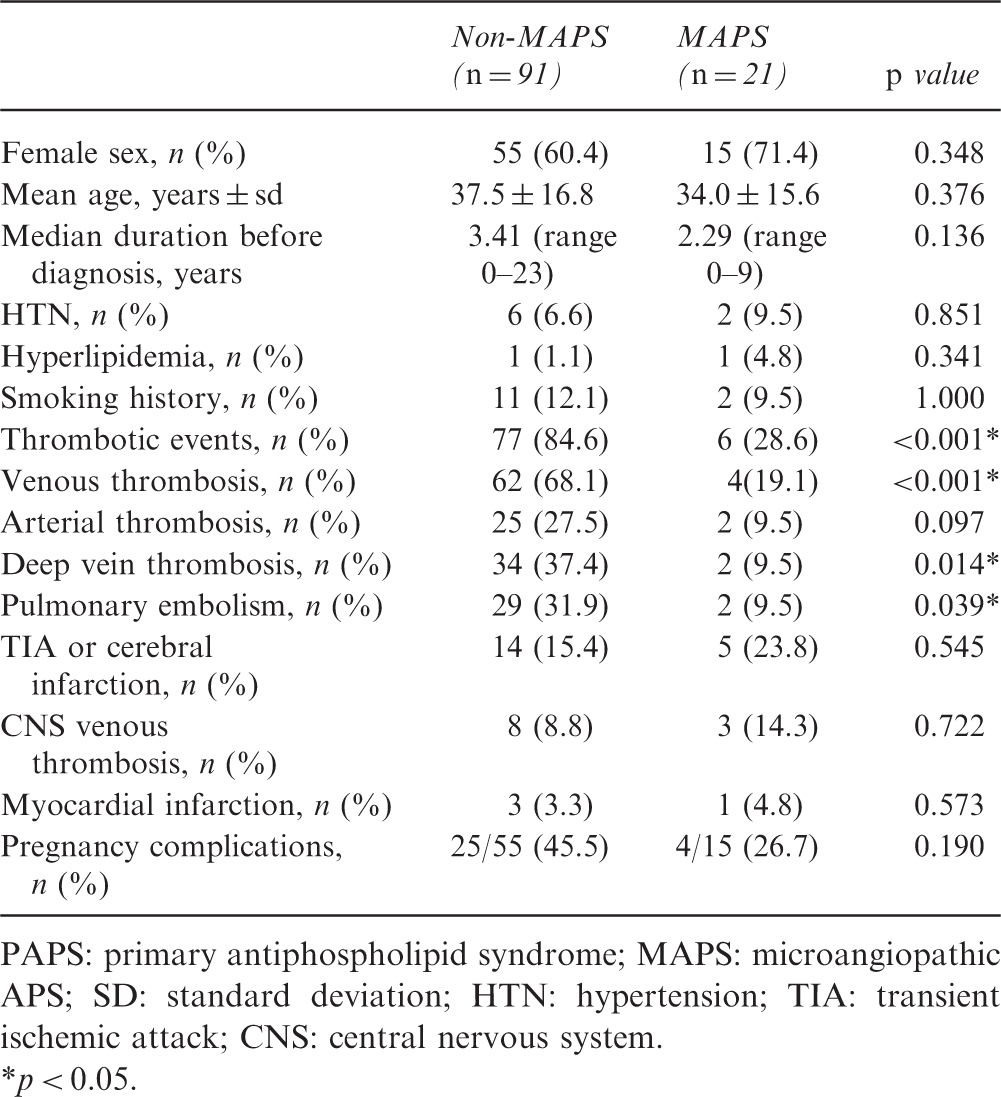
PAPS: primary antiphospholipid syndrome; MAPS: microangiopathic APS; SD: standard deviation; HTN: hypertension; TIA: transient ischemic attack; CNS: central nervous system.
p < 0.05.
The presence of arterial or venous thrombosis was remarkably lower in the MAPS group than in the non-MAPS group (28.6% vs. 84.6%, respectively, p < 0.001), especially venous thrombosis (19.1% vs. 68.1%, respectively, p < 0.001). In terms of major thrombotic manifestations, deep vein thrombosis (9.5% vs. 37.4%, p = 0.014) and pulmonary embolisms (9.5% vs. 31.9%, p = 0.039) were significantly more common in the non-MAPS group than in MAPS group. Transient ischemic attack or cerebral infarction (15.4% in non-APS vs. 23.8% in MAPS), cerebral venous thrombosis (8.8% in non-MAPS vs. 14.3% in MAPS), and myocardial infarction (3.3% in non-MAPS vs. 4.8% in MAPS) were also more common in the non-MAPS group, but these differences did not reach significance. As for pregnancy complications, no significant differences in the occurrence of obstetrical complications were observed between the two groups (45.5% in non-MAPS vs. 26.7% in MAPS).
We identified 21 patients with microvascular involvement in patients with PAPS, with a prevalence of 8.1%. Extra-criteria manifestations were also analyzed. The organ most affected was the skin (10 patients, including livedo reticularis, purpura, subcutaneous nodules, and superficial gangrene), followed by lung (three patients with alveolar hemorrhage), heart (one with heart valve disease and one with cardiomyopathy), and brain (ischemic encephalopathy with magnetic resonance imaging shown small cortical hypodensities). Seven patients had several concomitant manifestations at the same time.
Clinical and laboratory features
Laboratory features at baseline
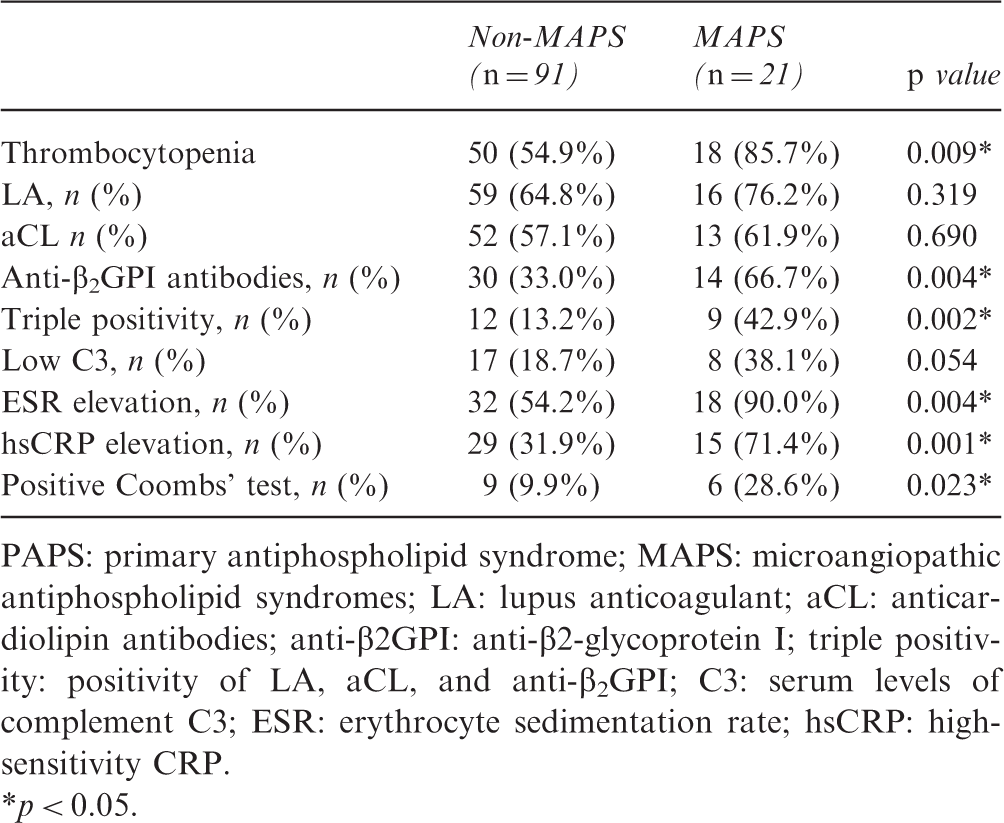
PAPS: primary antiphospholipid syndrome; MAPS: microangiopathic antiphospholipid syndromes; LA: lupus anticoagulant; aCL: anticardiolipin antibodies; anti-β2GPI: anti-β2-glycoprotein I; triple positivity: positivity of LA, aCL, and anti-β2GPI; C3: serum levels of complement C3; ESR: erythrocyte sedimentation rate; hsCRP: high-sensitivity CRP.
p < 0.05.
Multivariate statistical analysis and logistic regression
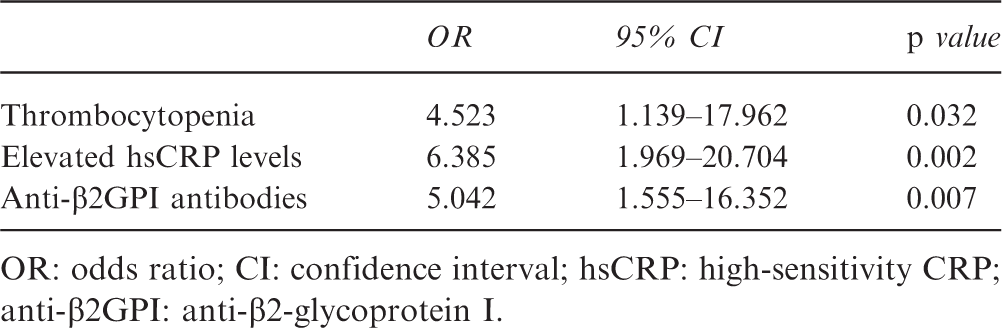
OR: odds ratio; CI: confidence interval; hsCRP: high-sensitivity CRP; anti-β2GPI: anti-β2-glycoprotein I.
In the multivariate analysis, associations remained significant for thrombocytopenia (odds ratio (OR) = 4.523, 95% confidence interval (CI) 1.139–17.962, p = 0.032), elevated hsCRP (OR = 6.385, 95% CI 1.969–20.704, p = 0.002), and positivity of anti-β2GPI antibodies (OR = 5.042, 95% CI 1.555–16.352, p = 0.007).
Treatment
Excluding patients whose treatment information was incomplete, 17 patients in the MAPS group and 89 patients in non-MAPS group were available for analysis. Nearly all patients received anticoagulation (warfarin or heparin) treatment, both in the MAPS group (n = 12, 60%) and non-MAPS group (n = 73, 80%), except for those who suffered from severe thrombocytopenia or comorbidities with a high risk of bleeding. Significantly more patients received aspirin and low molecular-weight heparin combination treatment in the MAPS group than in the non-MAPS group (52.9% vs. 11.2%, respectively, p = 0.001). Noticeably, 16 (94%) patients in the MAPS group received glucocorticoid treatment compared with 61 (68.5%) patients in the non-MAPS group. Glucocorticoids were used to treat severe thrombocytopenia, nephrotic syndrome, and autoimmune hemolytic anemia. Patients in the MAPS group had a remarkably higher probability of being treated with intravenous immunoglobulin than those in the non-MAPS group (47.1% vs. 9.0%, respectively, p < 0.001). Most of the patients in the MAPS group were administered a combination therapy, including two or more anticoagulant or antiplatelet therapies, immunosuppressive therapies, and intravenous immunoglobulin.
Survival analysis
Survival data were collected from the time of APS diagnosis until December 2016, with a median follow-up time of 10.0 years (interquartile range 2–7 years); the mean disease course was 53.5 ± 5.4 months. A total of 39 patients were lost during follow-up. The 1-, 3-, and 5-year survival rates did not differ between the two groups (95% in the MAPS group vs. 99% in the non-MAPS group). Regarding causes of death, patients with MAPS were more likely to die from direct causes attributed to thrombosis, such as pulmonary embolism (two deaths per 21 patients vs. one death per 91 patients) and stroke (three deaths per 21 patients vs. one death per 91 patients) than the non-MAPS group, respectively, but these differences did not reach statistical significance. Other causes of death included heart failure (no deaths vs. one death), multiple organ failure (three deaths vs. no deaths), acute kidney injury (two deaths vs. one death), and septic shock (no deaths vs. three deaths) in MAPS and non-MAPS groups, respectively.
To confirm the impact of MAPS on mortality, we compared cumulative survival rates between the two groups (Figure 1). A Kaplan–Meier analysis revealed a markedly higher all-cause mortality rate in MAPS group than in non-MAPS group (p = 0.0278; Figure 1). Additionally, in the MAPS group, most deaths occurred within the first year after MAPS onset, whereas the survival rate became stable in the non-MAPS group after the first year.
Kaplan–Meier analysis of survival in patients with primary APS.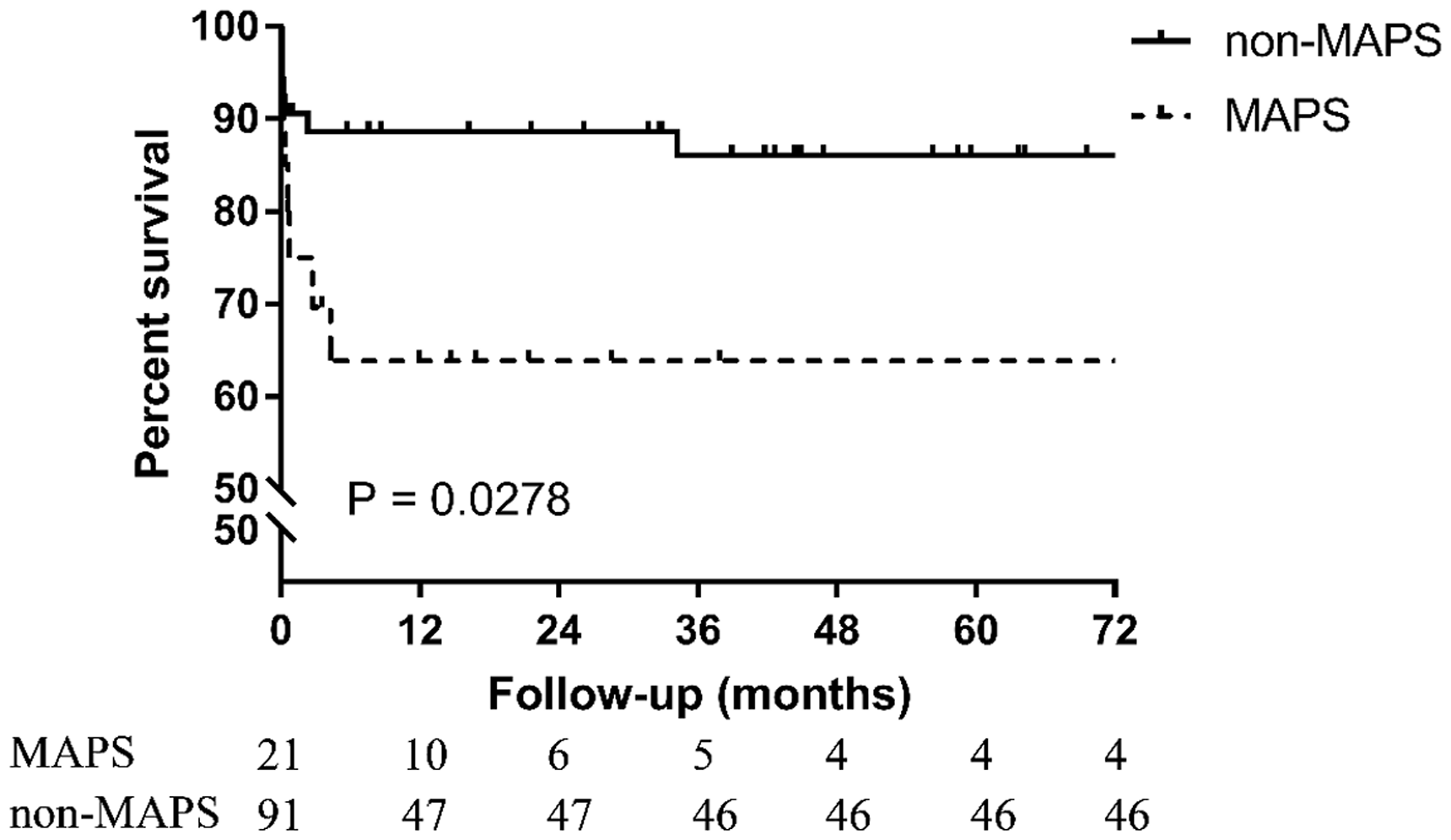
Discussion
In this study, we investigated the clinical and laboratory features of primary APS patients with microvascular involvement in a single-center, retrospective cohort. Clinically, APS-associated microvascular involvement was characterized by small-vessel occlusions and multi-organ involvement, whereas arterial or venous thromboses were less common. According to the multivariate analysis, thrombocytopenia, elevated hsCRP levels, and the presence of anti-β2GPI antibodies were independent risk factors for microvascular involvement in patients with primary APS. In terms of prognosis, patients with MAPS had a worse outcome than patients without MAPS.
The prevalence of elevated hsCRP levels in patients with MAPS suggests the existence of inflammation. Despite the historical view that inflammation plays an unimportant role in thrombogenesis in APS, increasing evidence shows that inflammation is responsible for the development of a procoagulant state.10,11 In CAPS, which is the most severe form of APS—the formation of acute, extensive, micro-vessel thromboses—researchers found that a group of cytokines (specifically, interleukin-1, interleukin-6, interferons, and tumor necrosis factor) were released in high amounts, which they referred to as the “cytokine storm.” 10 These cytokines are also known to cause a procoagulant state in systemic inflammatory response syndrome. 12 Recently, another inflammatory marker, ferritin, was proposed to play an important role in CAPS and has been included in a new group of syndromes termed “hyperferritinemic syndrome.” 13 In the light of such findings, inflammatory markers may serve as hallmarks of MAPS in patients with primary APS. In such conditions, inflammation control may improve outcomes.
In our study, anti-β2GPI antibodies were significantly more prevalent in the MAPS group than in the non-MAPS group, consistent with results from other studies. The CAPS registry, an international multicenter registry of CAPS, reported that the prevalence of the IgG and IgM isotypes of anti-β2GPI antibodies was 83.3% and 60.0%, respectively. 14 Another study of 114 patients with biopsy-proven lupus nephropathy found the prevalence of anti-β2GPI antibodies was higher in the TMA group than in the non-TMA group (p < 0.05). 15 Mechanisms by which anti-β2GPI antibodies induce microvascular occlusions are not completely understood. This process may involve phenotypic alterations of endothelial cells, circulating monocytes, and platelet activation. Results of in vitro studies have demonstrated that anti-β2GPI antibodies recognize, bind, and activate endothelial monolayers to upregulate adhesion molecules and proinflammatory cytokines, and stimulate chemokine secretion, inducing a proinflammatory, procoagulant phenotype. 16 Researchers also found that β2-glycoprotein inhibits von Willebrand factor-induced platelet aggregation, which can be neutralized by anti-β2GPI antibodies. 17 In CAPS, anti-β2GPI antibodies were found to trigger an endothelial cell signaling cascade as strong as that activated by lipopolysaccharide during sepsis. 11
The existence of hypocomplementemia in MAPS group suggests that complement activation may play a role in the development of MAPS. Consistent with our findings, Oku et al. found that slight hypocomplementemia was more prevalent in patients with primary APS than in those with other non-systemic lupus erythematosus connective tissue diseases as well as in healthy controls (16). A lower complement level was linked to higher anticoagulant activity in patients with primary APS. 18 Shamonki et al. showed that complement is deposited in the cytoplasm of trophoblasts in APS, which implies a correlation between pregnancy complications and complement deposition. 19 The use of eculizumab, a complement blocker, in patients with CAPS and multiple arterial thrombosis refractory to standard therapy has been successful. 20 One clinical trial is underway to assess the efficacy of eculizumab in preventing APS-associated TMA in patients with CAPS after kidney transplantation (ClinicalTrial.gov, NCT01029587).
Rates of thrombocytopenia were markedly higher in the MAPS group than in the non-MAPS group. A moderate decline in platelet number is a common feature of APS, with a reported prevalence ranging from 20% to 53%. 21 Thrombocytopenia was more frequently reported in patients with CAPS (63%) 22 and HELLP syndrome (94%). 23 Some researchers found the prevalence of thrombocytopenia was low (6%) in triple-positive patients and that platelet numbers dropped during the acute phase of CAPS. 24 This phenomenon implies the direct involvement of platelet consumption during the formation of a microthrombus in MAPS. For patients with consistent thrombocytopenia but no apparent evidence of elevated thrombus formation, some antiphospholipid antibodies, especially anti-β2GPI antibodies, may induce platelet aggregation, causing mild platelet consumption.25,26 Recently, a cross-sectional study investigated the relationship between circulating immune complexes formed by anti-β2GPI antibodies and extra-criteria manifestations in 57 APS patients. The presence of circulating immune complexes showed a strong association with thrombocytopenia (OR = 5.7) and complement consumption. 27
The survival rate of patients in the MAPS group dropped sharply in the first year after onset but recovered and remained stable afterwards. This finding suggests MAPS patients have a high mortality rate during the acute phase. However, this conclusion must be confirmed by further experiments with longer observation periods, larger sample sizes, and analyses of the causes of death. Early diagnoses and aggressive therapies are essential for MAPS management because of the high mortality in the early phase. According to the recommendations of the Task Force on CAPS, treatments should be directed at thrombotic events and the cytokine cascade, which typically involve a combination of anticoagulants, systemic glucocorticoids, plasma exchange, and intravenous immunoglobulin. 10
According to our analysis of 112 patients with primary APS, certain differences emerged that appear to distinguish MAPS patients from most patients with primary APS, supporting the idea that MAPS is an independent subset of APS. Because APS was first described in 1986, 28 small-vessel thromboses have been successfully documented in classic APS, but they do not dominate the clinical picture. 29 Asherson defined CAPS as a special subset of APS mainly affecting the small vessels of multiple organs. 30 Since then, an increasing number of cases have been characterized by TMA-like small-vessel occlusions, often affecting the kidney.4,5,9 Although the pathogenesis of APS nephropathy is not yet fully understood, antiphospholipid antibodies and complement activation have been recognized as important mechanisms in TMA development. 31 Other organs, such as the skin, central nervous system, lungs, heart, and eyes, may also be affected. 32 After reviewing numerous related reports, Levine et al. posted a list of APS clinical manifestations that likely result from microvascular occlusions. 33 Then 5 years later, Asheron et al. first defined the concept of MAPS. 2
A limitation of the present study was the comparatively small size of the patient population. This limitation made it impossible to assess the effects of medication because too few patients were on any one treatment regimen. Given the single-center, retrospective nature of data collection, there was possible selection bias. As a tertiary referral hospital, inpatients were seriously ill and may not adequately represent the general population in terms of APS characteristics. Additionally, despite our efforts to include qualified data, missing and insufficient information made it difficult to accurately identify all the medical data. For example, aPLs were used to report a total positivity in experimental reports and we could only use medical records to find out which isotype was positive. However, our study described for the first time the clinical characteristics and outcomes of primary APS with MAPS and compared it to classical APS. One of the advantages of this study was that the risk factors for thrombosis, such as hypertension, hyperlipidemia, and cigarette smoking, did not influence our findings because they were similar in both groups. Further, we only included patients with a primary APS diagnosis according to the Sydney criteria, excluding secondary APS, which may introduce confounding factors such as systemic lupus erythematosus.
Conclusions
In conclusion, APS-associated microvascular involvement is a newly defined but potentially life-threatening condition that requires high clinical awareness. Patients with this condition are characterized by manifestations of microangiopathy, with less arterial or venous thrombosis, elevated hsCRP levels, and low complement levels. Our study may shed light on future research on the potential disease mechanisms involved in MAPS.
Supplemental Material
LUP882506 Supplemental Material - Supplemental material for Clinical characteristics and risk factors of microvascular involvement in primary antiphospholipid syndrome: a longitudinal single-center study in China
Supplemental material, LUP882506 Supplemental Material for Clinical characteristics and risk factors of microvascular involvement in primary antiphospholipid syndrome: a longitudinal single-center study in China by Sun Y, J Zhao, P Zhang, C Wu, N Jiang, J Zhou, S Zhang, Q Wu, Q Wang, M Li and X Zeng in Lupus
Footnotes
Acknowledgement
Technical support from medical record department of Peking Union Medical College Hospital is gratefully acknowledged.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Chinese National Key Technology R&D Program, Ministry of Science and Technology (grant numbers 2017YFC0907601, 2017YFC0907602, and 2017YFC0907603), the Chinese National High Technology Research and Development Program, Ministry of Science and Technology (grant number 2012AA02A513), the Peking Union Medical College Hospital Fund for Distinguished Young Scholars (grant number JQ201706).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.