
Abstract
Myocardial infarction with non-obstructive coronary arteries (MINOCA) is a recently described, clinically significant entity, with prevalence rates ranging from 1% to 14% and a mean of 6% of all patients with myocardial infarction. Antiphospholipid syndrome (APS; Hughes syndrome) is characterized by the presence of antiphospholipid antibodies associated with thrombosis (arterial and/or venous) and/or pregnancy morbidity and could be the cause of MINOCA. Data on genetic predisposition to APS are scarce. The present study describes a unique case of monozygotic twin brothers who, at a young age, developed the same clinical presentation of APS. The diagnosis of APS was later confirmed, along with a diagnosis of systemic lupus erythematosus in one brother.
Background
The clinical presentation of the antiphospholipid syndrome (APS; Hughes syndrome) is not limited to vascular thrombosis or miscarriages, with the presence of antiphospholipid antibodies (aPL), such as anticardiolipin antibodies (aCL IgG/IgM), antiβ2 glycoprotein I antibodies (anti-β2GPI IgG/IgM) and a positive lupus anticoagulant test (LA).1,2 It includes a wide range of additional manifestations, including cardiovascular events. These cannot be explained solely by a thrombophilic state, and are not included in the classification criteria due to their low specifity. 3 Genetic factors are relevant in the development of APS. This has been demonstrated in animal models through familial occurrence of this syndrome and through its association with various human leucocyte antigen (HLA) alleles. 4
Myocardial infarction with non-obstructive coronary arteries (MINOCA) is a recently described, clinically significant entity, with prevalence rates ranges from 1% to 14% and a mean of 6% of all patients with myocardial infarction (MI). 4 It can be considered as a disease affecting either the epicardial arteries or the microcirculation, with various possible underlying pathophysiological mechanisms, including myocarditis, different prothrombotic diseases, as well as myocardial damage due to the discrepancy between myocardial demand and supply. 5
Case report
The present study describes a unique case of monozygotic twin brothers who, at a young age, developed the same clinical presentation of APS. The diagnosis of APS was later confirmed.
Brother A was admitted to hospital in October 2016 at 26 years of age with typical chest pain which had developed four days after an acute respiratory infection (tonsillopharyngitis) and seven days after the sudden death of his father. His monozygotic twin brother (brother B) had suffered the same type of event at the age of 18 (in 2008). Upon admission, brother A was obese (body mass index (BMI) 33.5 kg/m2), his glycaemia level was normal (4.5 mmol/L), as were the levels of total and low-density cholesterol (4.57 and 1.4 mmol/L, respectively), whereas his triglycerides were well above the normal range (4.7 mmol/L). His blood pressure was normal (120/85 mmHg), and he reported no prior instances of hypertension. Troponin I levels were elevated and they rose and fell during hospitalization, which is typical for acute myocardial injury (7.24, 16.06, < 0.01 µg/L). The level of CRP was high (103.1 mg/L). The electrocardiogram of brother A at admission revealed a sinus rhythm, with high T waves in the precordial leads, suggestive of early repolarization. A transthoracic echocardiography (TTE) exam at admission showed normal dimensions of the heart chambers, with global hypokinesia of the left ventricle (LV), grade I diastolic dysfunction and decreased systolic function, with LV ejection fraction (LVEF) estimated at 40–45%. Coronarography revealed no obstruction of the epicardial coronary arteries; there was no visualization of spasm or spontaneous dissection of the arteries. Bearing in mind that acute myocarditis might be the cause of MINOCA, cardiac magnetic resonance (CMR) was performed, and circular myocardial oedema in the medial and apical segments of the LV was detected, along with late gadolinium enhancement (LGE) in these segments (Figure 1). Pericarditis at the apical region was also described. The patient was discharged with the diagnosis of acute myocarditis and prescribed zofenopril 15 mg b.i.d., bisoprolol 2.5 mg q.d. and ibuprofen 400 mg t.i.d. In addition, endomyocardial biopsy (EMB) was scheduled at a different medical centre.
Three – dimensional retrospective image – based motion correction (TRIM) and late gadolinium enhancement sequences of CMR for brother A and brother B. Red arrows point to typical myocardial oedema.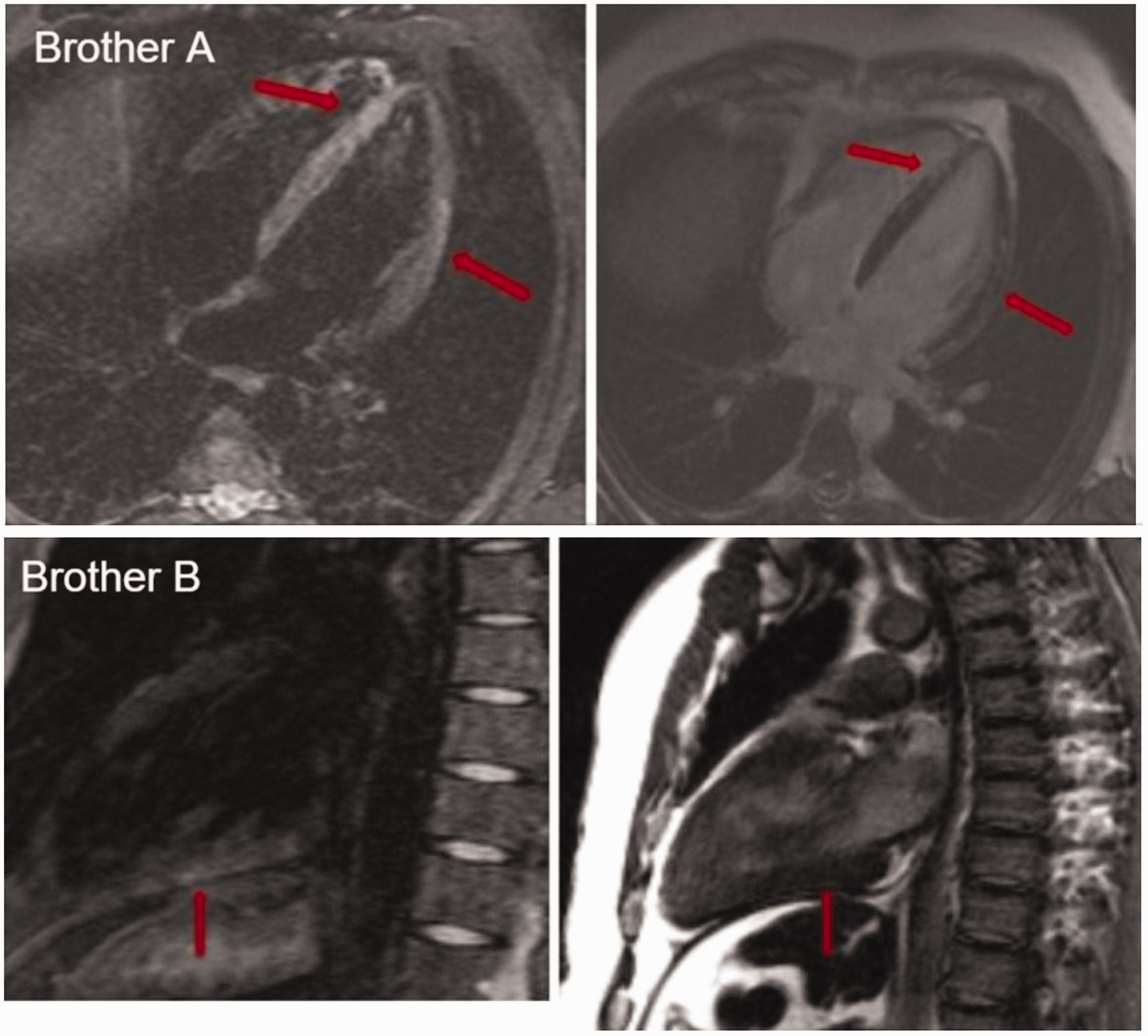
The following year (May 2017), at the age of 27, brother B was admitted to our cardiology department with the same clinical presentation (typical chest pain five days after acute tonsillopharyngitis, with ECG changes suggestive of early repolarization) and with dynamic changes in troponin T suggestive of acute MI (15.87; 0.18 µg/L). Of the standard atherosclerotic risk factors, only obesity (BMI 33.5 kg/m2), with normal levels of glycaemia, lipids and blood pressure, were reported. TTE showed a normal size of the heart chambers with no LV regional wall motion abnormalities, no impairment of LV relaxation and with LVEF estimated at 60%. Coronarography was not proposed. Since this was a repeated event of what had been diagnosed nine years before as myocarditis, with normal coronography findings and the same clinical course as brother A the year before, we suspected a thrombophilic state. CMR performed on brother B was also suggestive of myocarditis (Figure 1). The presence of LA was determined on the basis of two different screening tests: diluted activated partial thromboplastin time and sensitive activated partial thromboplastin time, as per the recommendations of the International Society of Thrombosis and Haemostasis. 6 LA in brother B was persistently positive (initial LA = 1.64, LA 12 weeks later = 1.55; normal range ≤ 1.2). The values of aCL (IgG/IgM) and anti-β2GPI (IgG/IgM) antibodies were measured by an ELISA (Binding Site) and expressed in G phospholipid (GPL) or M phospholipid (MPL) units (GPL-U and MPL-U), and were persistently negative. There were neither clinical nor serological features (negative antinuclear antibodies (ANA), dsDNA and extractable nuclear antigens, normal levels of serum complements) of either systemic lupus erythematosus (SLE) or any other autoimmune diseases. The patient was diagnosed with primary APS on the basis of the 2006 revised Sydney criteria, 7 and antiplatelet therapy (acetyl-salicylic acid 100 mg q.d.) with an anti-malaric drug (hydroxychloroquine 250 mg q.d.) was introduced. After the diagnosis of primary APS in brother B had been established, brother A was also tested. Repeated analysis of LA was positive (initial LA = 1.7, LA after 12 weeks = 2.36; normal range ≤ 1.2), whereas aCL (IgG/IgM) and anti-β2GPI (IgG/IgM) were negative, as in brother B. In contrast to brother B, brother A had a repeatedly high titre of homogenous type ANA (1:640). In keeping with the revised criteria of the American College of Rheumatology for SLE diagnosis, 8 based on the presence of CMR-proven myocardial and pericardial involvement, photosensitivity, non-erosive polyarthritis, low levels of serum complement and high levels of CRP and erythrocyte sedimentation rate, the diagnosis of SLE with associated APS was established. Analysis of anti-dsDNA and SSA/SSB antibodies was also performed, and they were negative. The same therapeutic regimen as in brother B (acetyl-salicylic acid 100 mg q.d. and hydroxychloroquine 250 mg q.d.) was introduced. On repeated TTE, no regional wall motion abnormalities were found, LVEF was estimated at 55% and no impairment of LV relaxation was detected. Repeated CMR in brother A revealed the persistence of LGE phenomena in the inferior and posterior walls of the LV but without indications for EMB.
Both brothers were tested for a panel of congenital thrombophilic states (more than six months after the event). There was no protein C, protein S, antithrombin deficiency or in vitro activated protein C resistance. The brothers were tested for genetic mutations: FII20210 prothrombin, FV Leiden, MTHFR C677T and PAI 4 G/5 G. Both of them tested positive only for the PAI 4 G/4 G mutation, which did not affect their treatment regimen.
Both brothers continue in a stable condition, with no repeated coronary events in further ambulatory follow-up or any symptoms or signs of heart failure.
Discussion
There are two main observations arising from this case report: one is the presence of genetic predisposition to APS, and the other is the importance of accurate and systematic diagnostic work-up for every MINOCA patient for the purpose of identifying its real cause and applying the most appropriate forms of treatment.
It was Exner et al. who, in 1980, first described three sets of siblings with LA, two of whom had more than one clinically affected member. 6 In a cohort of 577 European SLE patients, Galeazzi et al. described a significant correlation between aCL antibodies and HLA DR4 and DR7. 9 Although the genetic predisposition to APS, even in the setting of SLE, has been strongly confirmed, there has been no evidence of identical clinical presentation, as described in the present case. Ravindran et al. described a case of primary APS in monozygotic twins who were LA positive with high titres of aCL/IgG but with different clinical presentations. 10
The second ‘trouble’ in the present case study was the same clinical presentation, which was unfortunately repeated in brother B. According to the fourth universal definition of MI proposed by the European Society of Cardiology, the clinical definition of MI denotes the presence of acute myocardial injury detected by abnormal cardiac biomarkers when there is evidence of acute myocardial ischaemia. 11 This definition clearly distinguishes myocardial injury from MI, but also deals with the MINOCA diagnosis by stressing that MINOCA should be contemplated as a working diagnosis only, and that it requires additional diagnostics, including tests for thrombophilic states. In a VIRGO study, the MINOCA population of patients (11.1% of MI patients analysed) was significantly more predisposed to hypercoagulable states than MI patients with confirmed coronary artery disease (3.0% vs. 1.3%; p = 0.036). 12
As previously mentioned, up to 14% of MINOCA patients may have some thrombophilic state. 5 Bearing in mind that both brothers were diagnosed with APS at a young age, and for the purpose of proper prevention of future thrombotic events, the clinical decision was to test them for inherited thrombophilia. Both brothers were carriers of the homozygous mutation for PAI 4 G/4 G. Although this mutation is considered to be ‘weak’, it was important to be identified for the purpose of future monitoring of the health of these brothers and their offspring.
To conclude, genetic predisposition to APS and the precise detection of genes involved in its pathogenesis need confirmation in future studies. In clinical daily practice, this warrants establishing a protocol whereby aPL testing would be performed in all APS patients’ siblings. Also, aPL analysis should be performed in every MINOCA patient. Timely diagnosis of the pathogenesis would enable a proper therapeutic regimen, preventing repeated events.
Footnotes
Acknowledgements
We would like to thank Bojan Cakic, MD, Nebojsa Ninkovic, MD, Sasa Hinic, MD, and the entire staff of the Department of Cardiology at the University Hospital Center Bezanijska kosa in Belgrade, Serbia, for the treatment and diagnostics provided for both patients during their hospital stay.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: This work was supported by research grant number 175041 for 2011–2020 and by research grant number TR 32040 for 2011–2020, issued by the Ministry of Science of the Republic of Serbia.