
Abstract
Background
Low-density lipoprotein (LDL) levels are increased by proprotein convertase subtilisin kexin 9 (PCSK9) which targets the LDL receptor. We recently reported that PCSK9 ameliorates dendritic cell (DC) activation by oxidized LDL (OxLDL), which is abundant in atherosclerotic plaques and is also associated with cardiovascular disease (CVD) in systemic lupus erythematosus (SLE). Here, we investigated the role of PCSK9 in SLE.
Methods
PCSK9 levels were determined by ELISA among SLE patients (N = 109) and age- and sex-matched population-based controls (N = 91). Common carotid intima–media thickness (IMT) and plaque occurrence were determined by B-mode ultrasound. Plaques were graded by echogenicity. Human peripheral blood monocytes from SLE patients or controls were differentiated into DCs. The effects of PCSK9 and its inhibition by silencing were studied.
Results
PCSK9 levels were non-significantly higher among SLE-patients compared to controls but significantly associated with SLE disease activity, as determined by the Systemic Lupus Activity Measure (p = 0.020) or the SLE Disease Activity Index (p = 0.0178). There was no association between PCSK9 levels and atherosclerosis as determined by IMT, prevalence of plaques or echolucent (potentially vulnerable) plaques. PCSK9 levels were significantly associated with CVD among SLE patients but not after adjusting for age. OxLDL induced PCSK9 in DCs and DC maturation with increased expression of CD86 and HLA-DR. The effects were significantly stronger in DCs from SLE patients than from controls. Silencing of PCSK9 abolished OxLDL-induced DC maturation.
Conclusions
PCSK9 is associated with disease activity in SLE. One underlying cause could be OxLDL promoting DC activation which depends on PCSK9. OxLDL induces PCSK9 – an effect which is higher among SLE patients. PCSK9 could play an unexpected immunological role in SLE.
Keywords
Introduction
Systemic lupus erythematosus (SLE) can be described as a prototypic autoimmune disease. In addition to being an important clinical problem in itself, SLE is also of interest from other perspectives, related both to basic immunology and to cardiovascular disease (CVD), since the risk of CVD and atherosclerotic plaques is very high in SLE.1–5 Interestingly, atherosclerosis is nowadays seen as an inflammatory disease process.6,7 A combination of traditional and non-traditional risk factors for CVD and atherosclerosis have been implicated in SLE, which could account for the increased risk from an epidemiological point of view. 1
Among the traditional risk factors for CVD and atherosclerosis in SLE, hypertension and dyslipidaemia are important. The type of dyslipidaemia often present in SLE is interesting and is characterized by high triglycerides and low high-density lipoprotein, while low-density lipoprotein (LDL) is not necessarily high and can even be decreased in active disease. 8
There are also non-traditional novel and/or emerging risk markers, including antiphospholipid antibodies (aPL) which may cause both arterial and venous thrombosis but do not seem to be associated with atherosclerosis per se. Instead, other mechanisms seem to be of importance, such as competing out annexin A5 which has anti-inflammatory and anti-atherogenic properties. 9 Another example is natural antibodies against phosphorylcholine with potential protective properties.4,5,10
Atherosclerosis is characterized by the presence of activated immune-competent cells in the lesions, foam cells which have become inert and filled with oxidized LDL (OxLDL) and a necrotic core with dead cells.7,11 One common denominator in atherosclerosis, CVD and SLE is OxLDL, since this compound is increased and associated with CVD in SLE. 12
In a recent development, where both experimental and genetic studies were included, a new pathway of LDL metabolism was identified with proprotein convertase subtilisin kexin 9 (PCSK9). The genetic studies included missense variants, which led to low PCSK9 activity and a low risk of CVD,13,14 but other genetic variants leading to high PCSK9 activity and high levels of LDL have been reported. 15 This enzyme targets the LDL receptor and results in increased LDL levels. Inhibition of PCSK9 is now a novel treatment for CVD.16–18
We recently determined that OxLDL induces immune activation from human atherosclerotic plaque-derived T cells and dendritic cells (DC) from the same patient. 19 Both statins and PCSK9 inhibition could ameliorate these effects, though mechanisms including miRNA involvement were not identical and could thus potentially be immune modulatory.20,21
Here, we report that PCSK9 is associated with disease activity in SLE, and PCSK9 is important in OxLDL DC maturation in SLE. The implications are discussed.
Methods
The SLE Vascular Impact Cohort
The SLE Vascular Impact Cohort was studied herein, and has been described previously. 22 Briefly, in this study, 109 SLE patients from Karolinska University Hospital (Huddinge, Sweden) and 91 sex- and age-matched population-based controls were included. All patients fulfilled the 1982 revised criteria of the American College of Rheumatology for SLE. The study was approved by the Karolinska Institute Research Ethics Committee and was performed in accordance with the Declaration of Helsinki. All subjects gave informed consent before entering the study.
The study protocol included a written questionnaire, an interview, a physical examination by a rheumatologist, laboratory determinations and an ultrasound examination of carotid arteries. SLE activity was determined by use of the Systemic Lupus Activity Measure (SLAM)23,24 and SLE Disease Activity Index (SLEDAI).24,25 SLE organ damage was determined using the Systemic Lupus International Collaborating Clinics (SLICC) Damage Index.24,26 Blood samples were collected after an overnight fast between 07.30 and 10.00. Serum and cells were prepared separately in the laboratory and stored at –80°C.
Carotid B-mode ultrasonography
The ultrasonographic methods have been described in detail previously.27,28 Briefly, the right and left carotid arteries were examined with a duplex scanner (Sequoia; Siemens Acuson, Mountain View, CA) using a 6 MHz linear array transducer. 4 Two experienced ultrasonographers and physicians who were responsible for the ultrasonography were unaware of the patients’ and controls’ clinical data.
The far wall of the common carotid artery (CCA), 0.5–1.0 cm proximal to the beginning of the carotid bulb, was used for measurements of the intima–media thickness (IMT). The IMT was defined as the distance between the leading edge of the lumen-intima echo and the leading edge of the media-adventitia echo. The CCA lumen diameter was defined as the distance between the leading edge of the intima-lumen echo of the near wall and the leading edge of the lumen–intima echo of the far wall. 29 The mean values of the IMT and lumen diameter within the 10-mm-long section were calculated. When a plaque was observed in the region of the CCA measurements, the IMT was not measured.
Carotid plaque was defined as a localized intima–media thickening of >1 mm and at least a 100% increase in thickness compared to adjacent wall segments and was screened for in the common, internal and external carotid arteries. Plaque occurrence was scored as the absence of plaque, the presence of unilateral plaque and the presence of bilateral plaque. Plaque morphology in terms of echogenicity was assessed in a modified version of the classification proposed by Gray-Weale et al. 30 and graded from 1 to 4 as echolucent, predominantly echolucent, predominantly echogenic and echogenic. Echolucency was defined with the arterial lumen as reference and echogenicity with the far wall adventitia as reference.
Differences between repeated measures of IMT and lumen diameter were 4.9% and 2.4% (coefficient of variation), respectively (with an IMT of 0.44–1.02 mm and a lumen diameter of 4.38–7.9 mm). To determine the stretching effect of arterial distention secondary to increased arterial pressure on the thickness of the wall, we calculated the cross-sectional intima–media area by using the formula 3.14 ((lumen diameter/2 + IMT)2–(lumen diameter/2)2). Thus, the calculated intima–media area has been shown to be unaffected by artery distention variation in relation to blood pressure changes. 31 Furthermore, repeated plaque morphology classification showed a correlation coefficient of 0.7 (p < 0.05) between the first and second classification (n = 50).
PCSK9 levels in serum
PCSK9 levels in serum were measured by a standard ELISA according to the manufacturer’s instructions (R&D Systems, Abingdon, UK).
Generation of monocyte-derived DCs
Peripheral blood was sampled from SLE patients and case-control donors. DCs were generated essentially as described. 20 Briefly, peripheral blood mononuclear cells (PBMC) were isolated using Ficoll-Paque PLUS (GE Healthcare, Uppsala, Sweden). For monocyte isolation by adherence, 30 × 106 PBMCs in 10 mL RPMI/10% fetal calf serum were seeded in a Petri dish (10 cm diameter), and allowed to adhere for two hours in a 5% CO2 incubator at 37˚C. Non-adherent cells were removed, and the adherent cells were carefully washed twice with culture medium. Recovered monocytes were >90% pure as assessed by CD14 labelling. Monocytes were then differentiated to immature DCs over seven days, with 50 ng/mL human recombinant granulocyte-macrophage colony-stimulating factor and 25 ng/mL human recombinant interleukin-4 in RPMI 1640 supplemented with 2 mM glutamine, 10 mM HEPES and 1% penicillin/streptavidin. Copper-oxidized LDL from Source BioScience (Nottingham, UK) was used. For OxLDL treatment, an aliquot of immature DCs on day 6 were treated with OxLDL at 0.1, 1 or 10 µg/mL for 24 hours. For atorvastatin (AVA) treatment, an aliquot of DCs on day 5 were treated with 2 µM AVA for 24 hours and then stimulated with 2 µM AVA in the presence of 10 µg/mL OxLDL for another 24 hours. PCSK9 silencing was performed essentially as described. Briefly, DCs were transfected with the human PCSK9 Silencer or Silencer® Negative Control #1 siRNA (Thermo Fisher Scientific, Stockholm, Sweden). We used Lipofectamine® RNAiMAX transfection reagent (Invitrogen, Carlsbad, CA) at 20 nM concentration for six hours. After transfection, OxLDL and/or human recombinant PCSK9 (BioLegend, London, UK) were added. After this, cells were further incubated for 24 hours followed by functional study and RNA/protein analysis. 21 Cells were harvested and analyzed. Cell viability was >90%.
Flow cytometer analysis
DCs were collected and incubated with corresponding antibodies at 4˚C for 30 minutes. Anti-CD86-Percp/Cy5.5, CD83-APC, CD80-FITC (BD Biosciences, San Jose, CA) and HLA-II-PE (BioLegend, San Diego, CA) antibodies were applied for the staining. After washing, cells were analysed using a LSRII Fortessa™ cell analyser (BD Biosciences).
For PCSK9 intracellular staining in DCs, monensin (10 µg/mL; Sigma–Aldrich, St Louis, MO) was added into DC cultures for three hours before cell collection. After fixation with 2% formaldehyde and permeabilization in 0.5% saponin/phosphate-buffered saline, cells were incubated with anti-PCSK9 antibodies (Abbexa, Cambridge, UK) at 4˚C for 30 minutes. After washing, cells were incubated with 5% goat serum for 15 minutes followed by staining with APC-conjugated goat anti-rabbit IgG secondary antibody (Life Technologies Europe BV, Stockholm, Sweden) at 4˚C for 30 minutes. Cells were analysed using a LSRII Fortessa™ cell analyser (BD Biosciences) after washing, and data were analysed with FlowJo software. Quadrant markers were set accordingly to isotype-matched controls.
Quantitative real-time polymerase chain reaction
Total RNA was extracted using the RNeasy Mini Kit (Qiagen, Sollentuna, Sweden), and cDNA was reversely transcribed using Superscript II (Gibco-BRL) according to the manufacturer’s instructions. Quantitative real-time polymerase chain reaction (qRT-PCR) was performed for selected genes. We tested the gene PCSK9 (Hs00545399_m1; Applied Biosystems, Foster City, CA). The expression level of the housekeeping gene GAPDH (Hs02758991_g1) was used to normalize for differences in input cDNA. qRT-PCR was carried out using TaqMan gene expression pre-synthesized reagents and master mix (Applied Biosystems) using a 7500 real-time PCR system (Applied Biosystems). The expression ratio was calculated using the ΔΔCT method.
Statistical analysis
The correlation between PCSK9 levels was performed at the baseline level for all subjects to investigate the association between PXCS9 and SLAM, SLEDAI and SLICC. As our data were skewed, we used Spearman correlation. Comparison of PCSK9 levels between cases and controls and for prevalence of atherosclerotic plaques was done by analysis of variance after logarithmic transformation of data. These analyses were performed using SAS v9.4 (SAS Institute, Cary, NC). In experimental studies where the role of PCSK9 on immune activation was investigated, we used Student’s t-test. For all statistical analyses, a two-tailed p-value of <0.05 was considered significant. p-Values are presented for individual experiments (*p < 0.05, **p < 0.01).
Results
Clinical associations
Basic information about the study group is presented in Table 1. PCSK9 levels did not differ significantly between SLE patients and controls (Table 1 and Figure 1). High levels of PCSK9 was more common in SLE (e.g. above the 25th and 33rd percentiles), but these differences, like mean levels, were not statistically significant (data not shown).
Confounding factors of disease and control groups.
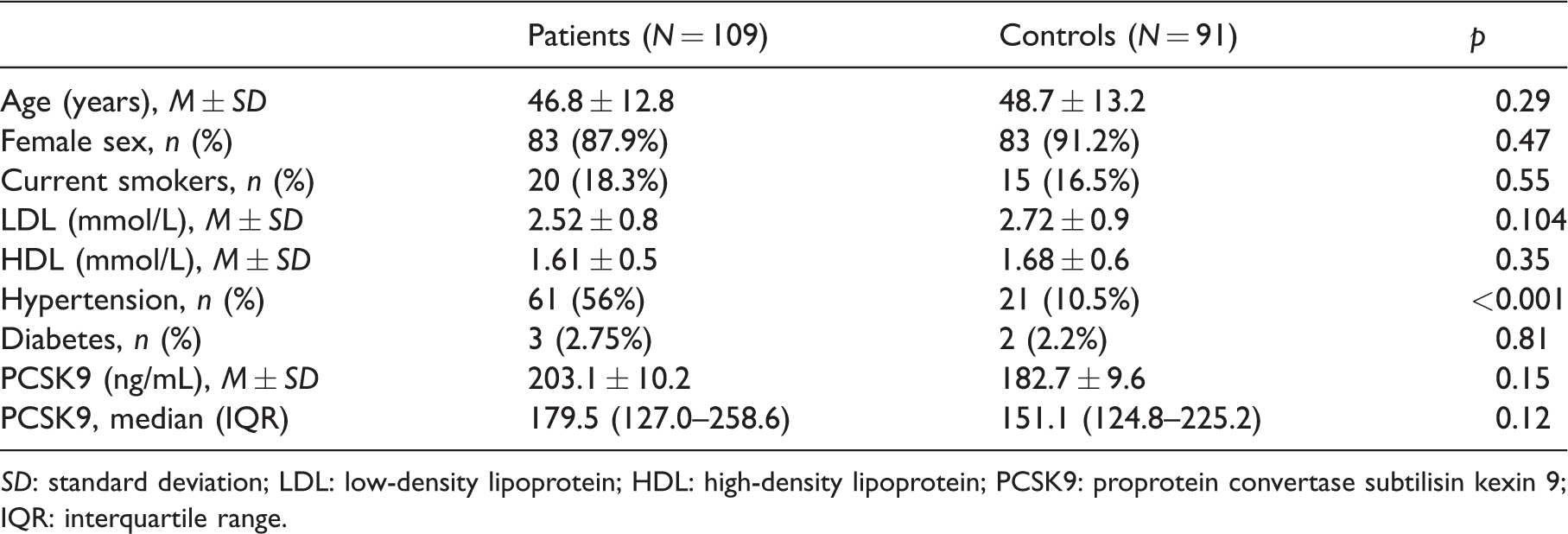
SD: standard deviation; LDL: low-density lipoprotein; HDL: high-density lipoprotein; PCSK9: proprotein convertase subtilisin kexin 9; IQR: interquartile range.
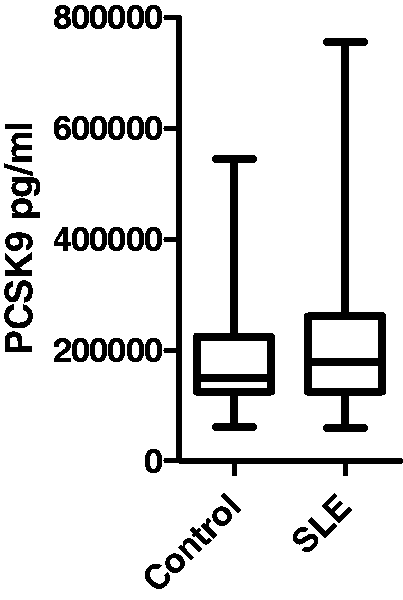
Proprotein convertase subtilisin kexin 9 (PCSK9) levels among cases and controls as determined by ELISA.
Median values among controls were 151.01 ng/mL (interquartile range (IQR) 100.4 ng/mL) and among cases 179.6 ng/mL (IQR 131.7 ng/mL). Both median and IQR are given. Interquartile range is being is equal to difference between 75th and 25th percentile.
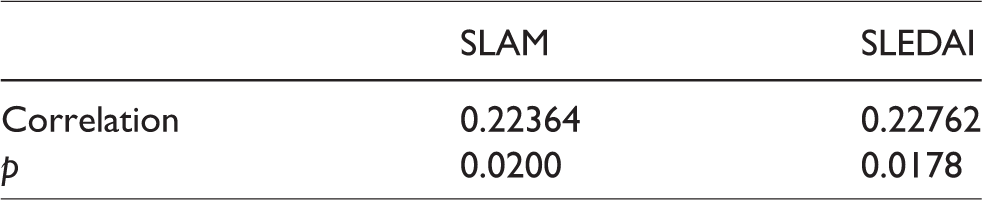
Among SLE patients, PCSK9 levels were significantly and positively associated with disease activity as determined by either the SLAM or SLEDAI (Table 2). There was no association with disease damage (SLICC).
Positive association of PCSK9 levels with SLAM and SLEDAI.
There were no significant associations between PCSK9 levels and atherosclerotic plaques or echolucent (potentially vulnerable) plaques or with IMT in SLE or with medications as corticosteroids or hydrochlorochine (data not shown).
PCSK9 levels were significantly associated with CVD in SLE. If the highest quartile was compared to the lower quartiles, the odds ratio (OR) was 9.95 (95% confidence interval (CI) 1.20–78) but when adjusted for age, this association did not reach statistical significance (OR = 5.69; 95% CI 0.69–47). There was no significant association between LDL levels and PCSK9 levels in SLE (data not shown).
DCs from SLE patients were more sensitive to OxLDL-induced activation
DCs were treated with various doses of OxLDL at 0.1, 1 and 10 µg/mL. In line with our previous findings, 21 the expression of co-stimulatory molecules CD80, CD83, CD86 and HLA-DR were enhanced in OxLDL (10 µg/mL)-treated DCs. Interestingly, these expressions on DCs from SLE patients (SLE-DCs) were already promoted by treatment of OxLDL at 1 µg/mL, but not on DCs from control subjects (Ctl-DCs; Figure 2).
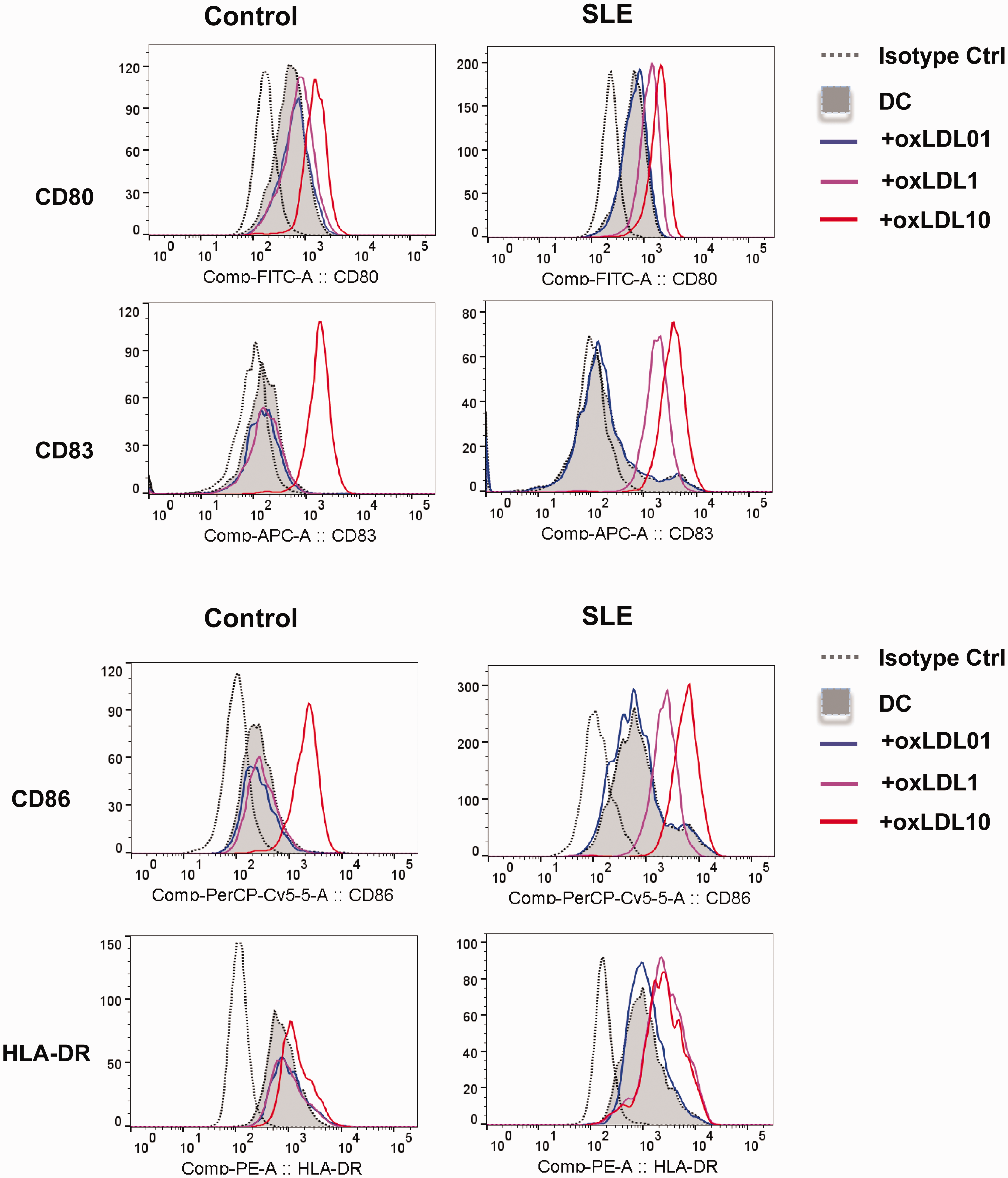
Expression of CD80, CD86, CD83 and HLA-DR. On day 7, dendritic cells (DCs) were analysed by flow cytometry. FACS plots are representative of four independent experiments. Results in staple diagrams represent the mean fluorescence intensity (MFI) ± standard deviation (SD) of four experiments. **p < 0.01, DC + oxidized low-density lipoprotein (OxLDL-1) vs. DC; **p < 0.01, DC + OxLDL-10 vs. DC.
SLE-DC patients expressed higher PCSK9, and OxLDL treatment increased the expression
PCSK9 gene expression in DCs was analysed by RT-PCR (Figure 3). Interestingly, the expression of PCSK9 in SLE-DCs was higher than it was in Ctl-DCs (2.08 ± 0.23 vs 1.01 ± 0.27, n = 8, p < 0.001; Figure 3A). Treatment of OxLDL substantially induced PCSK9 expression both in SLE-DCs (16.54 ± 5.87 vs. 2.08 ± 0.23, n = 8, p = 0.0027) and in Ctl-DCs (5.71 ± 1.54 vs. 1.01 ± 0.27, n = 8, p = 0.0088). PCSK9 expression in SLE-DCs was already enhanced by treatment of OxLDL at 1 µg/mL (5.12 ± 1.71 vs. 2.18 ± 0.16, n = 3, p = 0.0162) but not in Ctl-DCs. Treatment of AVA did not affect the induction of PCSK9 by OxLDL.
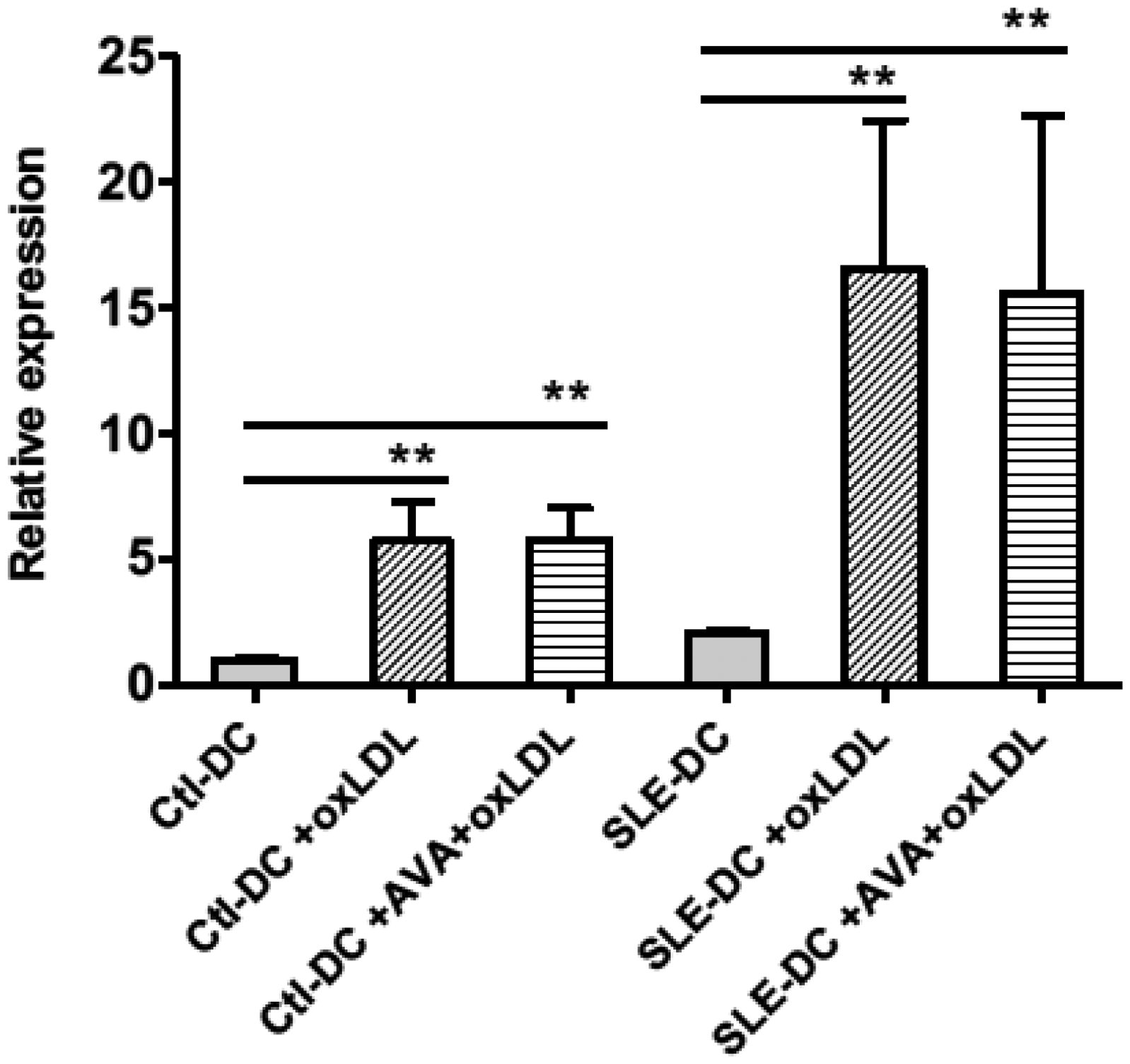
PCSK9 was induced in OxLDL-treated DCs. On day 6, DCs were treated with OxLDL at 0.1, 1 and 10 µg/mL for 24 hours. For atorvastatin (AVA) treatment, an aliquot of DCs on day 5 were treated with 2 µM AVA for 24 hours and then stimulated with 2 µM AVA in the presence of 10 µg/mL OxLDL for another 24 hours. Expression of PCSK9 in DCs was tested by quantitative real-time polymerase chain reaction. The value was normalized as fold change to that of non-treated DC samples. Results represent the mean±SD of eight experiments for controls and three for SLE patients. *p<0.05; **p<0.01.
PCSK9 played an essential role in SLE-DC maturation induced by OxLDL
To study the potential role of PCSK9, we downregulated its expression in DCs from SLE-patients with PCSK9 silencer. By treatment with PCSK9 silencer, the OxLDL-induced up-regulation of co-stimulatory molecules CD80, CD83, CD86 and HLA-DR was repressed (Figure 4).
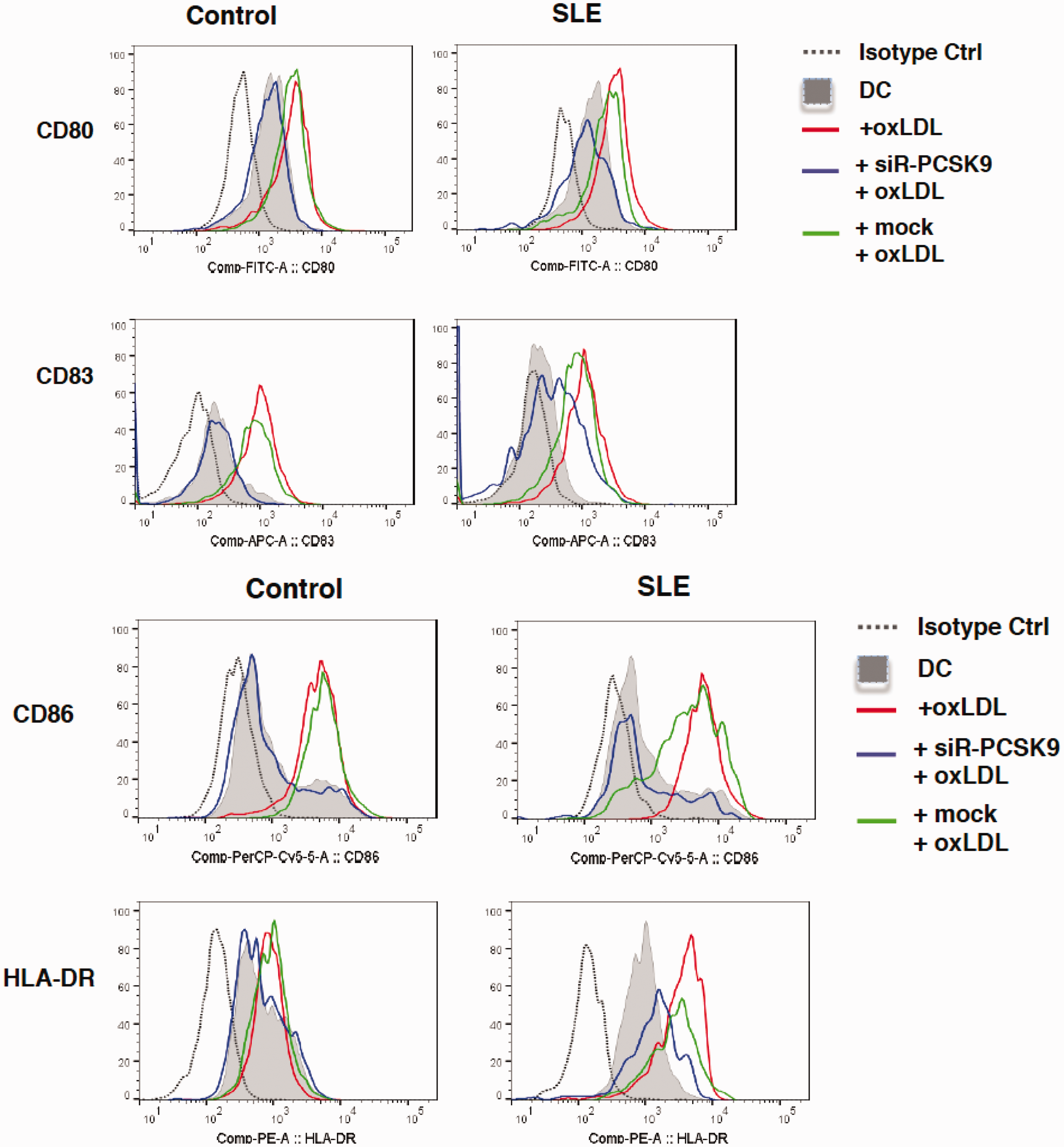
PCSK9 silencing abolishes OxLDL-induced SLE-DC activation and maturation. On day 6, DCs were treated with 10 μg/mL OxLDL for 24 hours. For down-regulation of PCSK9, PCSK9 silencer (siR-PCSK9) or negative control siRNA (mock) was transfected into DCs with Lipofectamine® RNAiMAX in a 20 nM concentration on day 6. OxLDL was added six hours after transfection, and further incubated for 24 hours. Expression of CD80, CD86, CD83 and HLA-DR. On day 7, DCs were analysed by flow cytometry.
Discussion
Here, we report that PCSK9 levels are associated with disease activity in SLE as determined by both the SLEDAI and SLAM. Even though the levels were higher among SLE cases compared to controls, this difference was not statistically significant. There were no associations with the prevalence of atherosclerotic plaques or echolucent (and potentially vulnerable) plaques. Even though PCSK9 levels were strongly associated with CVD, this association did not hold when adjusted for age. It is still possible that one reason for the strong association between age and CVD is related to PCSK9, and thus the importance of it is unclear and deserves further study in larger cohorts. Even though not associated with the disease per se, our findings thus suggest that PCSK9 still could play a role in inflammation typical of SLE, since there was an association with disease activity. In contrast, there was no association with LDL levels.
We are aware of one previous study on PCSK9 and SLE. In that study, the authors reported that levels of PCSK9 were raised in SLE, and were associated with atherosclerosis and nephritis (and also atherosclerosis as determined by IMT measures) in SLE. 32 We thus cannot confirm these findings, since we did not determine significantly raised levels of PCSK9 in SLE.
Another difference with the cited study is thus that we could not detect any associations with PCSK9 levels and measures of atherosclerosis, measured in detail by ultrasound and including both prevalence of plaques and quality of plaques, where echolucency could be a sign of vulnerability. 4
There could be several reasons which could explain these discrepancies. Importantly, the patients in our study were much older (mean age = 48 years vs. 33 years) and appear to have a less severe type of disease, with the SLAM and SLEDAI being lower (median value for SLEDAI on our study was 24 while it was 6 in the other study). 32 Since PCSK9 levels in our findings were associated with disease activity, it is thus possible that differences in both age and disease activity between these two studies could explain the reported discrepancies between the two studies on PCSK9 levels in SLE. In line with this are the higher values of PCSK9 in the other study. There could also be differences in selection of controls (which in our study were population based). Further studies are needed to shed light on the clinical role of PCSK9 levels in SLE and its role as risk marker for SLE compared to controls. Importantly, the role of PCSK9 prospectively must also be scrutinized, studies of which are under way in our laboratory.
We then studied potential underlying mechanisms which could provide an explanation of the association between disease activity in SLE and PSCK9 levels. We focused on two previous findings from our group. One is that OxLDL is raised in SLE and associated with CVD (and disease activity in SLE). 12 Another is our recent report that OxLDL can induce immune activation in T cells from atherosclerotic plaques and DCs from the same individual, that inhibitors include Annexin A5 and further that heat shock protein 60 is involved in the process. 19 To complicate things further, both statins and PCSK9 inhibition by silencing have unexpected immunological effects in relation to OxLDL-induced immune activation, which are not related to the LDL targeting properties of these compounds. An anti-inflammatory phenotype was promoted among T cells, with increases in T regulatory cells, and DC activation and maturation was also inhibited. However, underlying mechanisms were not identical, with differences in miRNA usage among others.20,21
Here, we confirm our previous findings that OxLDL promotes activation and maturation in DCs from control individuals (in the previous report, we used PBMCs from Buffy coats from healthy blood donors as controls), and also demonstrate that DCs from SLE patients are more sensitive to OxLDL-induced activation and maturation. Such activated DCs could in principle promote disease activity in SLE. This is also of interest, since, as mentioned, circulating OxLDL is increased in SLE. 12 OxLDL in SLE could thus be one underlying factor behind inflammation in SLE, even more so since OxLDL effects on DC maturation were stronger than among controls.
Another interesting finding is that DCs from SLE patients expressed more PCSK9 compared to controls. Even though PCSK9 is mainly produced in the liver, it is also expressed in other tissues, including the vascular wall.18,33 One example of an anti-inflammatory effect not directly related to lipid lowering is that PCSK9 can have local effects, increasing inflammation in atherosclerotic chimeric mice, expressing PCSK9 in macrophages. 34 DCs overexpressing PCSK9 could thus in principle also promote local inflammation in atherosclerotic plaques and other tissues.
Statins are widely used to lower LDL and to decrease the risk of subsequent myocardial infarction and other types of CVD. Statins have long been known to have pleiotropic anti-inflammatory effects based on interference with prenylation and thus on cytokine production which could contribute to a more general anti-inflammatory effect, and thus based on what could be called a side effect of the inhibition of HMG-CoA-reductase. Such anti-inflammatory effects have been much discussed, but it is not clear what role they play in explaining the positive clinical effects of statins on the risk of CVD.10,20 It is interesting to note that statin use is not associated with a decreased risk of developing SLE. 35 Further, statin therapy does not decrease disease activity in SLE as determined by the SLEDAI. 36 Here, we demonstrated that statins did not inhibit OxLDL-induced PCSK9 expression. If PCSK9 plays a role in SLE, our finding could potentially contribute to the lack of effect of statins on disease activity in SLE.
The prevalence of atherosclerotic plaques is raised in SLE, 4 and the risk of CVD is very high in this disease.1,3 Further, CVD in SLE is associated with atherosclerosis in this disease condition, even though there is also an increased risk of thrombosis, for example through aPL, which also play a role. 1
DCs are professional antigen-presenting cells, orchestrating cell-mediated immune responses through different mechanisms, including cytokine signalling and, they also play an important role, for example, in efferocytosis. In atherosclerotic plaques, DCs are prevalent, and are believed to play an important role in different stages of development – early but also late when plaques become complicated and may rupture, leading to CVD. DCs and T cells co-localize in sites of plaque rupture.37–40 DCs could also play an important role in SLE. Aberrant DC function has been reported in many publications, and mature and activated DCs are a feature of increased disease activity in SLE. 41 DC activation by OxLDL and also with PCSK9 as a player could thus play an important role in both disease conditions, and these findings could also provide yet another link between them.
A limitation with the experimental studies on DCs herein is that a relatively small number of patients were tested in the ex vivo systems, and larger studies are needed to establish the effect and characterize them more in detail. Likewise, larger cohort studies, also with follow-up data, are needed to establish the role of PCSK9 as risk marker and potential risk factor in SLE.
Taken together, our findings indicate that PCSK9 could play an important and also unexpected role in SLE. First, it is associated with disease activity, and since OxLDL is raised in SLE, it induces expression of PCSK9 and also maturation in DCs from SLE patients. Silencing of PCSK9 abolishes the effects, and it is thus possible that PCSK9 inhibition could be beneficial to patients with SLE, especially in patients with increased disease activity.
Conclusions
Here, we report for the first time that PCSK9 is associated with disease activity in SLE. One underlying cause could be OxLDL, which is raised in SLE and promotes DC activation, more so among SLE patients than controls. This effect is PCSK9 dependent. PCSK9 could thus play an unexpected immunological role in SLE.
Footnotes
Acknowledgements
We thank Tomas Jogestrand and Thomas Gustafsson for the ultrasound analyses of the carotid arteries, and Max Vikström for help with statistical analyses.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship and/or publication of this article: This study was done with support from Swedish Heart and Lung Foundation, Swedish Association against Rheumatism and Amgen (unconditional grant). The granting agencies had no influence on the study design or interpretation.