
Abstract
Objective
To compare the frequency of rare variants in genes of the pathophysiologically relevant endosomal Toll-like receptor (eTLR) pathway and any quantifiable differences in variant rarity, predicted deleteriousness, or molecular proximity in patients with systemic lupus erythematosus (SLE) and healthy controls.
Patients and methods
65 genes associated with the eTLR pathway were identified by literature search and pathway analysis. Using next generation sequencing techniques, these were compared in two randomised cohorts of patients with SLE (n = 114 and n = 113) with 197 healthy controls. Genetically determined ethnicity was used to normalise minor allele frequencies (MAF) for the identified genetic variants and these were then compared by their frequency: rare (MAF < 0.005), uncommon (MAF 0.005–0.02), and common (MAF >0.02). This was compared to the results for 65 randomly selected genes.
Results
Patients with SLE are more likely to carry a rare nonsynonymous variant affecting proteins within the eTLR pathway than healthy controls. Furthermore, individuals with SLE are more likely to have multiple rare variants in this pathway. There were no differences in rarity, Combined Annotation Dependent Depletion (CADD) score, or molecular proximity for rare eTLR pathway variants.
Conclusions
Rare non-synonymous variants are enriched in patients with SLE in the eTLR pathway. This supports the hypothesis that SLE arises from several rare variants of relatively large effect rather than many common variants of small effect.
Keywords
Introduction
Systemic lupus erythematosus (SLE) is the prototypic systemic autoimmune disease with a heterogenous array of clinical phenotypes and organ involvement characterised by deposition of circulating immune complexes in various body tissues. Whilst almost all components of the immune system have been implicated in the pathogenesis of SLE, one of the most consistent immunologic features is a prominent type-1 interferon (T1-IFN) signature driven by activation of the endosomal Toll-like receptor (eTLR) pathway within plasmacytoid dendritic cells (pDC).1,2 This enhanced T1-IFN, and the subsequent autoimmunity, arises from a complex interplay of environmental and genetic risk factors. Next generation sequencing (NGS) techniques have permitted discovery of multiple new disease mechanisms and have further developed our understanding of the factors that contribute to an individual’s genetic risk of SLE. 3
It is well established that SLE has a compelling genetic basis as evidenced by strong twin-twin concordance and familial clustering.3,4 Several hypotheses have been proposed to model the genetic basis of sporadic SLE. Genome-wide association studies (GWAS) have identified common genetic variants with modest risks for the development of several autoimmune diseases, including SLE. 5 Very few of these alleles have discernible phenotypic effects when tested experimentally, and thus it has been proposed that it is the cumulative and epistatic effect of these alleles that predisposes to autoimmunity. However, one major limitation to this approach is that rare variants are typically missed or underemphasised by conventional GWAS. 6 With the increased uptake of NGS it has become recognised that rare (minor allele frequency (MAF)<0.005) and novel alleles have substantially greater phenotypic effects than common alleles across many conditions.7–9 Indeed, autosomal dominant and recessive forms of SLE exist as the result of novel and ultrarare variants.10,11 Together, from these observations it has been proposed that common alleles associated with SLE by GWAS may mark regions containing rare and more deleterious genetic variants.12,13 Recently we demonstrated that rare and novel missense variants in GWAS-identified SLE risk genes BLK and BANK1 had functional consequences leading to augmented T1-IFN responses. 14
Dendritic cells are critical for both the initiation of adaptive immune responses and the maintenance of self-tolerance. 15 pDCs are professional antigen presenting cells with the greatest T1-IFN producing capacity within the immune system. 16 The endosomal Toll-like receptor (eTLR) pathway is a major driver of T1-IFN production in response to viral infection. 17 Nucleic acids, including viral RNA and DNA, but importantly self-nucleic acids, and other ligands, are capable of activating the eTLR system through interaction with TLR7/8 and TLR9. The pDC T1-IFN response has been shown to be important in the development of lupus-like autoimmunity in murine models and human disease. 18 The activation of murine pDCs and subsequent activation of B cells by T1-IFN leads to a break in self-tolerance and the induction of antinuclear antibodies. 19
It is therefore unsurprising that given the prominent role of T1-IFN and pDCs, genes with essential roles in T1-IFN production and eTLR signalling are frequently implicated by GWAS in SLE development.20,21 As a result we hypothesized that, given the pathophysiologically important role of the eTLR pathway, 22 pre-existing GWAS association of many eTLR genes, and increasing recognition of the role of rare variants, patients with SLE may have a greater burden of rare variants in the eTLR system.
Patients and methods
Human recruitment
Written informed consent was obtained as part of the Australian Point Mutation in Systemic Lupus Erythematosus study (APOSLE), the Centre for Personalised Immunology and Genetic Mutation (GEM) in Kidney Disease program. The study was approved by and complies with all relevant ethical regulations of the Australian National University and ACT Health Human Ethics Committees (Protocol Reference: ACT Health ETH.01.15.015). Recruitment of patients with SLE occurred in Australia in two phases: between 2008 and 2011 and between 2015 and 2017. Initially, individuals with SLE were recruited after meeting the American College of Rheumatology criteria for SLE. Already recruited patients were then audited in 2015 against the Systemic Lupus International Collaborating Clinics (SLICC) classification criteria 23 and only patients meeting the SLICC classification criteria for SLE were considered in this analysis. The 45 and Up 24 and ASPREE datasets 25 were used as reference healthy controls, accessed through the MGRB Collaborative (http://sgc.garvan.org.au/mgrb/initiatives).
Next generation sequencing
Saliva was collected in Oragene™ DNA self-collection kits and purified using PrepIT™ DNA purification kits (Oragene) and treated with Ribonuclease A (Qiagen Cat# 19101). DNA samples were enriched with Human SureSelect XT2 All Exon V4 Kit and sequenced by Illumina HiSeq 2000 (Illumina, Inc.). WES had 21% low or uncovered exon bases compared with 4% low or uncovered exon bases for WGS. Bioinformatic analysis was performed at JCSMR, ANU. Raw sequence reads were aligned to the reference genome (Hg19) and single-nucleotide variants and small insertions and deletions called using GATK.
Statistical modelling
The possibility for confounding of minor allelic frequencies (MAF) by ethnic differences in frequency has been previously demonstrated. 26 To overcome this we used the available genetic sequencing data to determine the ethnicity of each individual in both SLE cohorts and the healthy controls using GEMTools.27,28 These were grouped into 6 separate ethnic clusters in gnomAD (African, American, East Asian, Finnish, non-Finnish European, and South East Asian). Admixture was corrected for by the use of the rADMIXTURE algorithm 29 applied on the Dodecad K7b ancestry dataset. 30 GEMTools clustering was then used to identify each sample’s ethnically-matched population frequency in gnomAD. The obtained MAFs were normalised for each variant using the individual’s genetically-determined ethnicity. We then controlled for the effect of genetic admixture on the burden of variants.
The remaining variants were then divided into common (MAF >0.02), uncommon (MAF 0.005-0.02), and rare (MAF <0.005) variants. Intronic variants obtained from the WGS of the elderly healthy controls were discarded and only exonic variants were included.
The burden of genetic variants in the eTLR pathway was assessed by comparing the extent to which individuals from the three cohorts had two or more variants in genes associated with the eTLR pathway.
To exclude an effect from differences in sequencing technique, we compared overall exome coverage comparing the median total number of exonic variants per individual in the SLE cohorts and the healthy control cohort.
Statistical analysis
Where multiple comparisons of the differences between median values were required, the Kruskal-Wallis test was used. Where this identified statistical significance between the cohorts, the differences between the medians of two groups were compared using the Mann-Whitney U Test. The difference between the burden of genetic variants was compared using a Chi-square test.
Results
Increased burden of rare endosomal Toll-like receptor variants in SLE patients
227 patients with SLE who had undergone whole exome sequencing (WES) were randomly sorted into an initial cohort of 114 patients (SLE 1) and a replication cohort of 113 individuals (SLE 2). The two cohorts were compared with 197 healthy controls (HC) recruited as part of the Medical Genome Reference Bank (MGRB) cohort who had undergone whole genome sequencing (WGS). 31 Overall, there were 9993 exonic variants in the genes of the eTLR pathway in the HC group and 12090 exonic variants in the combined SLE cohorts (6072 variants in SLE 1 and 6018 variants in SLE 2).
We first compared the overall frequency of nonsynonymous and synonymous variants in 65 eTLR genes (Table S1) between the three cohorts. There was no significant difference observed in the overall median number of nonsynonymous eTLR gene variants in SLE 1 (23.0, IQR: 3.3) and SLE 2 (22.0, IQR: 4.0) compared with the HC cohorts (22.0, IQR: 4.0, ANOVA p = 0.08) (Figure 1(a)). Rare genetic variants are increasingly recognised to have larger phenotypic effects than their more common counterparts. 32 We therefore hypothesised that although the overall burden of nonsynonymous eTLR variants was similar, the burden of rare non-synonymous eTLR variants would be greater in patients with SLE compared to HC. Indeed, we observed a higher median number of rare non-synonymous eTLR variants in patients with SLE 1 (1.0, IQR: 1.0, p = 0.0002) and SLE 2 (1.0, IQR: 1.0, p = 0.0493) compared to HC (0.0, IQR: 1.0) (Figure 1(b)). There was no difference in the frequency of rare synonymous variants suggesting this difference was specific to nonsynonymous variants and not an artefact of sequencing between cohorts (Figure 1(b)).
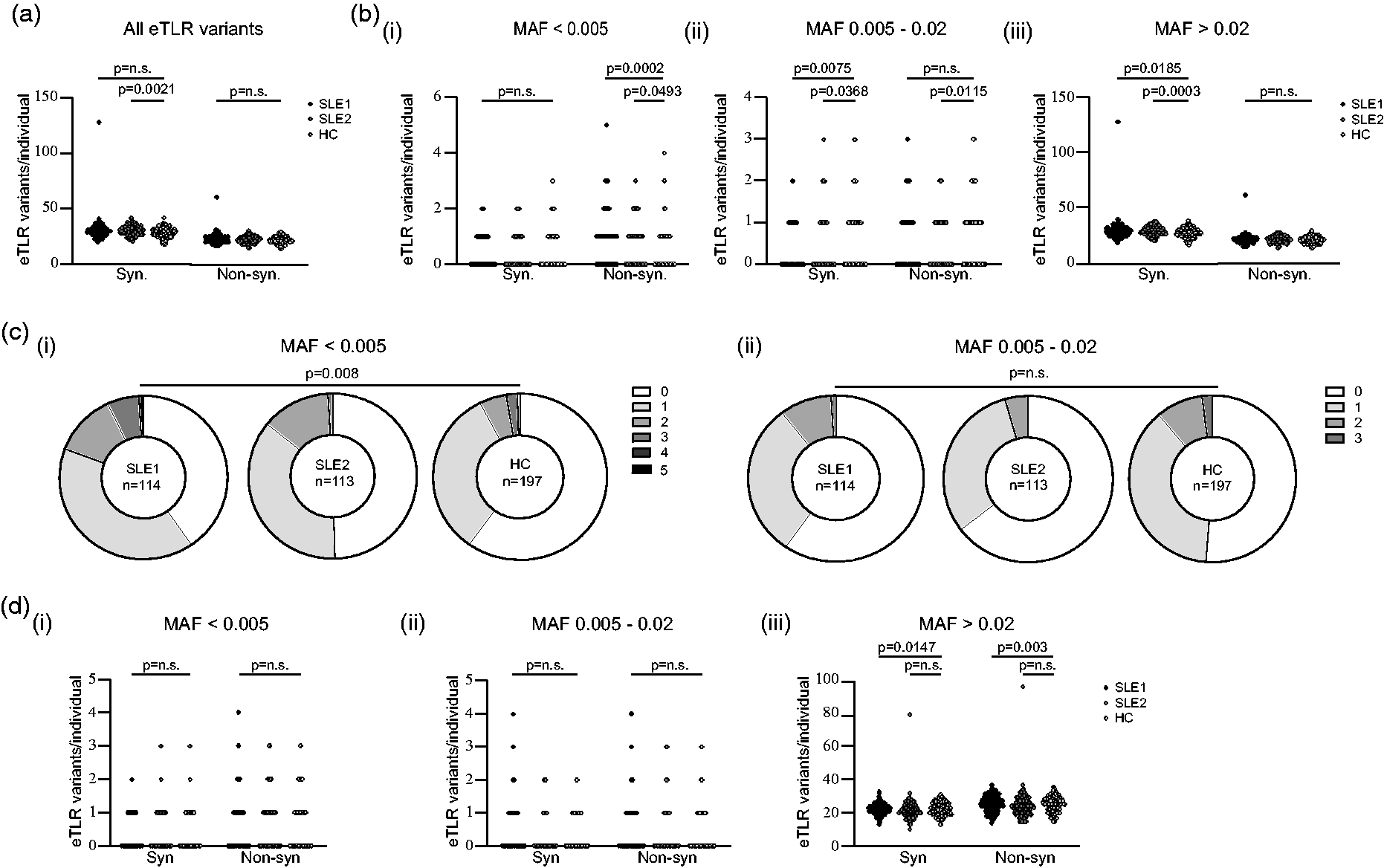
SLE patients have a greater burden of rare non-synonymous variants in genes within the eTLR pathway but not in a set of randomly selected genes. (a) The total number of synonymous and non-synonymous variants per individual in an initial SLE cohort (SLE 1, n = 114) and SLE replication cohort (SLE 2, n = 113) and healthy control cohort (HC, n = 197) in genes associated with the eTLR pathway. (b) The number of (i) rare (MAF <0.005), (ii) uncommon (MAF 0.005–0.02) and (iii) common (MAF >0.02), synonymous and non-synonymous variants per individual in eTLR pathway genes in SLE 1, SLE 2 and HC cohorts. (c) The burden of non-synonymous rare (MAF <0.005) and uncommon (MAF 0.005–0.02) variants in the eTLR pathway across the SLE 1, SLE 2 and HC cohorts. (d) The number of synonymous and non-synonymous variants per individual in SLE 1, SLE 2 and HC cohorts in 65 randomly selected genes for (i) rare (MAF <0.005), (ii) uncommon (MAF 0.005–0.02) and (iii) common (MAF >0.02) variants.
While rare (MAF < 0.005) non-synonymous eTLR variants are more common in people with SLE, as the allelic frequency of the non-synonymous eTLR increased, this increase was not present when more common non-synonymous variants were examined. Specifically, for non-synonymous eTLR with an MAF of 0.005–0.02, there was a higher median number of nonsynonymous eTLR variants in HC compared to SLE 2 (p = 0.01) and a trend towards a higher median number compared to SLE 1 (p = 0.2, Figure 1(b)). Furthermore, there were no significant differences in common nonsynonymous eTLR variants (Figure 1(b)).
If the observed increase in rare nonsynonymous eTLR variants is specific for this pathophysiologically important pathway, we hypothesised that there should not be increases in nonsynonymous variants in genes unrelated to the eTLR pathway. We randomly selected 65 genes (Table S2) and compared the frequency of rare variants between cohorts. No increase in the number of non-synonymous or synonymous rare variants was detected (Figure 1(d) and S1B, ANOVA p = 0.8). Together this suggests that there is an increase in the prevalence of rare nonsynonymous variants in SLE patients which is specific to the eTLR pathway.
We hypothesised that the greater frequency of rare eTLR variants in the SLE cohort may reflect a greater burden of eTLR variants for each individual patient. We compared the number of individuals in each cohort by number of eTLR variants. There were significantly greater numbers of individuals with 2 or more rare non-synonymous eTLR variants in SLE 1 and SLE 2 cohorts compared to HC (Figure 1(c), p = 0.008), but this was not replicated for uncommon (MAF 0.005–0.02) non-synonymous eTLR variants. There was no difference in the number of individuals with 2 or more non-synonymous variants in the randomly selected genes (Fig S1, p = 0.8). Together, these data indicate that rare non-synonymous variants in the pathophysiologically important eTLR pathway are enriched in SLE patients.
We postulated that there may be qualitative differences in eTLR variants in SLE patients compared to HC. Rarity is associated with greater phenotypic effects, and so we compared the allele frequency of eTLR variants, but observed no difference in the median allele frequency of rare or common variants between the SLE and HC cohorts (Figure 2(a), ANOVA p = 1.0 and 0.7, respectively). There was a lower median MAF in the uncommon variants of the SLE 2 cohort relative to healthy controls (p = 0.02) but this was not present when compared to the SLE 1 cohort (p = 0.9) and may reflect the small sample size. The Combined Annotation Dependent Depletion (CADD) score provides a quantifiable bioinformatic score to predict the deleteriousness of variants. We compared CADD scores for all non-synonymous eTLR variants according to MAF category, however, there were no significant differences in CADD scores between rare variants (Figure 2(b), ANOVA p = 0.8). The median CADD score for common variants was significantly higher among healthy controls than for the SLE cohorts (p < 0.0001 for both). Furthermore, there was a statistically greater median CADD score for uncommon variants in the healthy controls compared to the SLE 2 cohort (p = 0.0001). The significance of this finding was not clear. The findings of a higher median CADD score for HC in common variants was also identified in the analysis of 65 random genes (Fig S2, p < 0.0001 for both SLE cohorts). We initially hypothesised that this was the result of different ethnic mixtures between the cohorts leading to segregation of different common alleles. When CADD scores greater than zero for non-synonymous eTLR genes were selected for ethnically matched non-Finnish Europeans, there was no significant difference in CADD scores between the two groups (Tables S3A and S3B). Therefore, the increased median CADD scores in the HC cohort in both the eTLR and random genes was due to an increased number of low CADD score variants in the SLE cohorts.
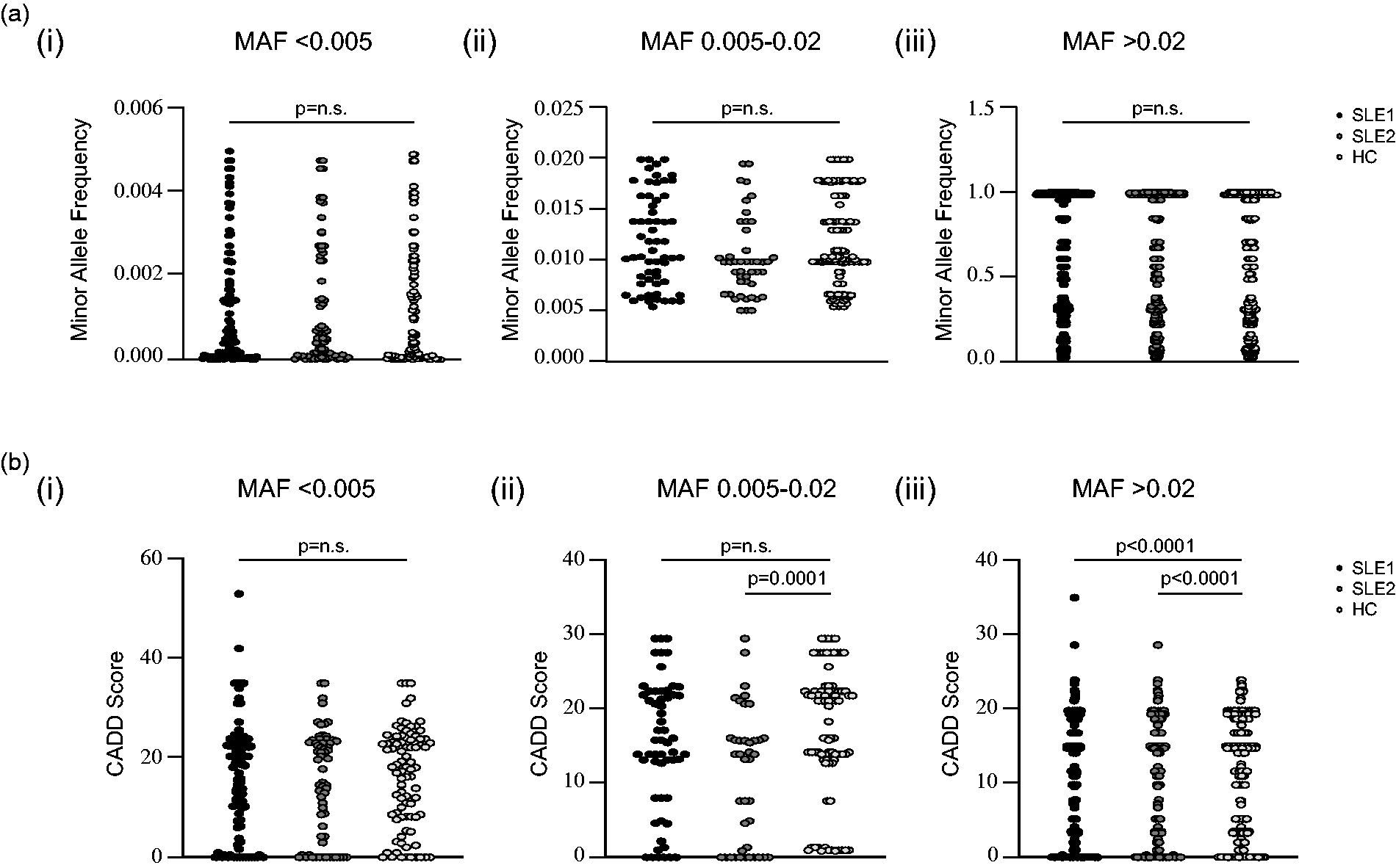
Comparison of minor allele frequency and CADD scores for variants in genes associated with the eTLR pathway. (a) The minor allele frequencies (MAF) for (i) rare (MAF <0.005), (ii) uncommon (MAF 0.005–0.02) and (iii) common (MAF >0.02) non-synonymous variants in genes associated with the eTLR pathway in the SLE 1, SLE 2 and HC cohorts. (b) CADD scores for (i) rare (MAF <0.005), (ii) uncommon (MAF 0.005–0.02) and (iii) common (MAF >0.02) non-synonymous variants in genes associated with the eTLR pathway in the SLE 1, SLE 2 and HC cohorts.
It has been hypothesised that molecular proximity of interacting gene variants may exert epistatic effects. We hypothesised that SLE patients may not only have a greater burden of rare variants, but also that these variants may be in close functional proximity and therefore potentially have epistatic effects predisposing to SLE. For individuals with 2 or more non-synonymous variants, we identified the median genomic distance and degree of separation between these using the Human Gene Connectome.33,34 We did not observe a difference in genomic distance (Figure 3(a), ANOVA p = 0.8) nor degree of separation (Figure 3(b), ANOVA p = 0.6) between SLE patients and HC. Thus, we concluded that we could not detect qualitative differences in molecular proximity between the non-synonymous variants in genes of the eTLR pathway through bioinformatic means with the limited number of patients available for this study.
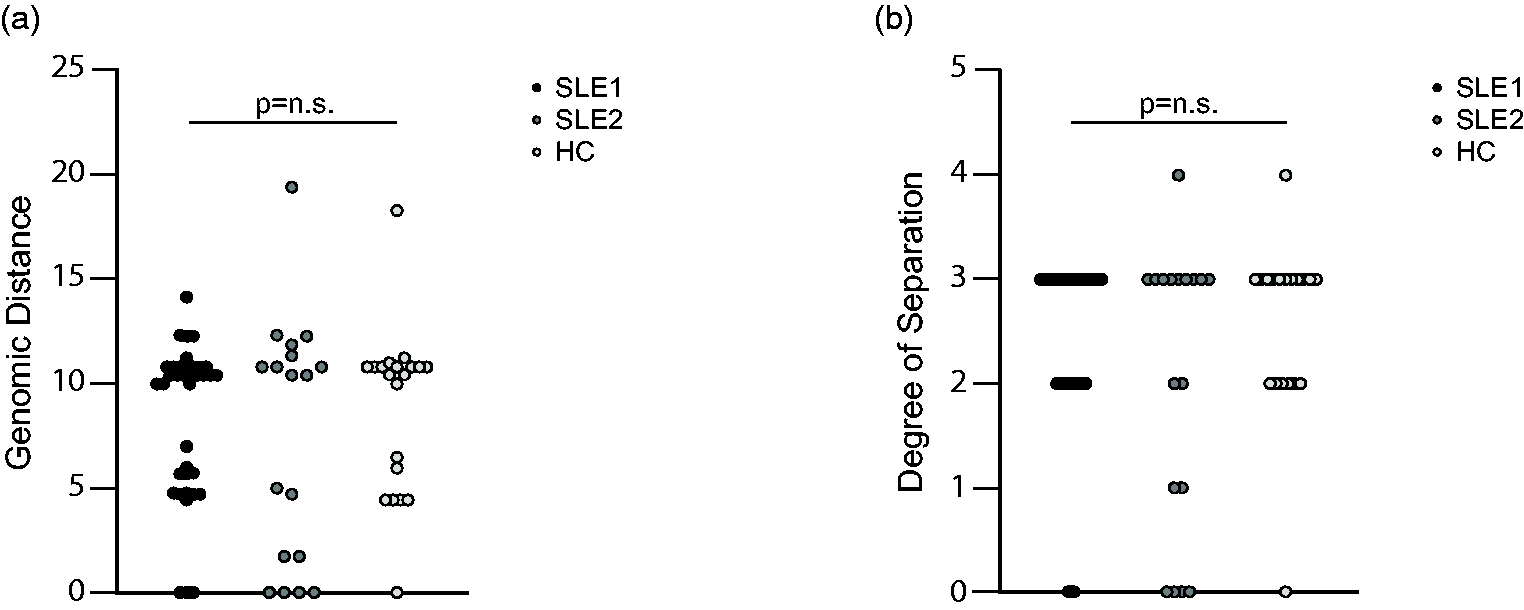
The molecular proximity of variants in the eTLR pathway in individuals with 2 or more rare (MAF <0.005) variants. (a) The genomic distance for the SLE 1, SLE 2 and HC cohorts. (b) The degree of separation for the SLE 1, SLE 2 and HC cohorts.
Discussion
We have demonstrated an increased number of rare nonsynonymous variants affecting the eTLR system in patients with SLE compared with healthy controls. This finding supports the emerging evidence that rare variants in pathophysiologically significant pathways may influence risk for the development of SLE. To date, limited studies have explored the role of rare variants in an unbiased manner but have not explored these variants in relevant pathways. We propose that the combined assessment of genes within relevant disease pathways may help further elucidate the genetic underpinning of sporadic SLE.
The importance of the T1-IFN pathway in SLE development and propagation is well recognised with several active attempts at novel therapy development targeting this pathway.35,36 This T1-IFN signature is stimulated via activation of the endosomal Toll-like receptor pathway within pDCs.1,2 pDCs are known to be capable of breaking immune tolerance and initiating autoimmune responses through this signalling.37,38
Genetic predisposition is a prominent contributor to the development of SLE. There is increasing recognition of new potential disease pathways through next generation sequencing and approaches such as GWAS. However, GWAS usually identifies high-frequency variants which often have limited impact on protein function or lie in non-coding regions. 39 The results of GWAS studies typically identify common SNPs of modest effect that, even when considered together, only account for a small proportion of genetic inheritance. 40 This is in keeping with the “omnigenic model” of complex genetic traits which states that common variants in peripheral genes outside of core disease-related genes, but with interrelated pathways, accounts for most of genetic predisposition to disease. 41 This has then generated the hypothesis that SLE results from multiple high-frequency, weak variants that combine to produce a highly “polygenic” compound genetic injury that leads to the development of disease.
However, rare variants have also been associated with the development of disease, 14 and at the extreme of this paradigm are examples of monogenic forms of SLE and interferonopathies. Our alternative hypothesis is that rare variants of relatively large effect, that have been explored in other conditions, 42 explain the “missing hereditability” in SLE. These are variants that arise in core disease-related genes and contribute strongly to genetic risk of disease, assist a detailed understanding of disease pathogenesis and identify biologic pathways that may be amenable to targeted therapeutic intervention. However, rare genetic variants are more likely to be population-specific and thus it is essential to correct for genetically determined ethnicity. 43
In this study we show that SLE patients have a higher burden of rare variants in the eTLR pathway and are more likely to have a “compound injury” with more than one variant in the pathways. This increased prevalence supports an “oligogenic” mechanism for the development of SLE in which several rare variants, perhaps across different relevant functional pathways, combine to cause a compound injury leading to disease, rather than the multiple common variants proposed in the GWAS model of genetic disease development. A role for rare variants in the development of phenotypic differences has been demonstrated in other instances. 7
Together these results suggest that rare genetic variants in the eTLR pathway are more common in patients with SLE. This pattern is what would be predicted with lower frequency variants being more injurious than high frequency variants that are seen commonly.
This study is limited by the small size of the cohorts and warrants further assessment with a larger cohort of patients. Another limitation relates to differences in sequencing platform between the patients of the SLE and HC cohorts. Total exome coverage is higher in WGS than WES which should have favoured increased variants in the HC cohort relative to the SLE cohorts, although we could not detect significant differences in variants counts across cohorts.
In conclusion, this study demonstrates that patients with SLE have a higher median number of rare nonsynonymous variants in the eTLR pathway compared to healthy controls. This was not seen in a random gene selection. There was also a higher burden of rare variants in the eTLR pathway in patients with SLE compared to healthy controls, although we could not find evidence of increased molecular proximity of variants in individuals with multiple eTLR variants.
Overall, this study demonstrates that rare nonsynonymous variants in the eTLR pathway are enriched in individuals with SLE. This supports the contribution of rare variants in disease-related pathways towards the development of SLE. Larger studies of the eTLR pathway, and other SLE-related pathways, with additional experimental testing of variants may help to better understand the complex genetic basis of SLE.
Supplemental Material
sj-pdf-1-lup-10.1177_09612033211033979 - Supplemental material for Increased burden of rare variants in genes of the endosomal Toll-like receptor pathway in patients with systemic lupus erythematosus
Supplemental material, sj-pdf-1-lup-10.1177_09612033211033979 for Increased burden of rare variants in genes of the endosomal Toll-like receptor pathway in patients with systemic lupus erythematosus by Tom N Lea-Henry, Aaron Chuah, Maurice Stanley, Vicki Athanasopoulos, Malcolm R Starkey, Daniel Christiadi, A Richard Kitching, Matthew C Cook, Thomas D Andrews, Carola G Vinuesa, Giles D Walters and Simon H Jiang in Lupus
Supplemental Material
sj-pdf-2-lup-10.1177_09612033211033979 - Supplemental material for Increased burden of rare variants in genes of the endosomal Toll-like receptor pathway in patients with systemic lupus erythematosus
Supplemental material, sj-pdf-2-lup-10.1177_09612033211033979 for Increased burden of rare variants in genes of the endosomal Toll-like receptor pathway in patients with systemic lupus erythematosus by Tom N Lea-Henry, Aaron Chuah, Maurice Stanley, Vicki Athanasopoulos, Malcolm R Starkey, Daniel Christiadi, A Richard Kitching, Matthew C Cook, Thomas D Andrews, Carola G Vinuesa, Giles D Walters and Simon H Jiang in Lupus
Supplemental Material
sj-pdf-3-lup-10.1177_09612033211033979 - Supplemental material for Increased burden of rare variants in genes of the endosomal Toll-like receptor pathway in patients with systemic lupus erythematosus
Supplemental material, sj-pdf-3-lup-10.1177_09612033211033979 for Increased burden of rare variants in genes of the endosomal Toll-like receptor pathway in patients with systemic lupus erythematosus by Tom N Lea-Henry, Aaron Chuah, Maurice Stanley, Vicki Athanasopoulos, Malcolm R Starkey, Daniel Christiadi, A Richard Kitching, Matthew C Cook, Thomas D Andrews, Carola G Vinuesa, Giles D Walters and Simon H Jiang in Lupus
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: SHJ is supported by RACP Jacquot NHMRC Award for Excellence, Jacquot Research Establishment grant and NHMRC project grants. TLH is supported by a Jacquot Research Entry scholarship.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.