
Abstract
Background
Intestinal and hepatic manifestations of lupus seem to be underestimated in comparison to other major organ lesions. Although recent data point to gut-liver axis involvement in lupus, gut permeability dysfunction and liver inflammation need to be more investigated.
Objective
This study aims to assess fecal calprotectin, intestinal tight junction proteins and liver inflammation pathway in wild-type murine imiquimod- induced lupus.
Methods
C57BL/6 mice were topically treated on their right ears with 1.25 mg of 5% imiquimod cream, three times per week for six weeks. Fecal calprotectin was collected at day 0, 22 and 45. Renal, liver and intestinal pathology, as well as inflammatory markers, intestinal tight junction proteins, and E. coli protein in liver were assessed at sacrifice.
Results
At six weeks, lupus nephritis was confirmed on histopathology and NGAL and KIM-1 expression. Calprotectin rise started at day 22 and persists at day 45. Protein expression of Claudine, ZO-1 and occludin was significantly decreased. E. coli protein was significantly increased in liver with necro-inflammation and increased TLR4, TLR7, and pNFκB/NFκB liver expression.
Conclusion
This study is the first to demonstrate early fecal calprotectin increase and liver activation of TLR4- NFκB pathway in wild-type murine imiquimod-induced lupus.
Introduction
Lupus is a prototype of multi-organ auto-immune diseases. Although gastrointestinal symptoms are reported to occur in more than 50% of lupus patients at some point during their disease, these manifestations attract far less attention than the other major organ involvements and seem to be underestimated. 1 Several studies have used lupus-prone mice to elucidate the pathogenesis of lupus, but there is a very few models of inducible lupus in wild-type mice except pristane-induced lupus and the graft-versus-host reaction. 2 Recently an inducible lupus model that uses epicutaneous application of TLR7 agonists (like imiquimod) has been described in several genetic backgrounds. This model leads to several phenotypic and functional changes characteristic of human lupus, including antibodies development and renal immune complex deposition, but also interestingly liver inflammation with mononuclear cell infiltrates around the portal veins.2,3 Although lupus hepatitis reports were recently surveyed in a systematic review of human series, liver inflammation is not constantly documented in lupus, and animal models studies are starting to explore its different pathways and its relation to intestinal permeability dysfunction. 1
Moreover, lupus-related gut permeability is gaining interest in lupus pathogenesis investigations. 4 The intestinal epithelial barrier, with its intercellular tight junctions, controls the equilibrium between tolerance and immunity to non-self-antigens. 1 Tight junctions dysfunction with intestinal permeability disruption seems to be a primary defect in many auto-immune diseases, such as Crohn's disease, celiac disease, ankylosing spondylitis, Behçet disease, type 1 diabetes mellitus and primary biliary cirrhosis.5–8 The loss of protective function of mucosal barriers that interact with the environment seems to be a key element in autoimmunity development, in addition to genetic predisposition and exposure to triggering non-self-antigens. 9
The gut of lupus-prone mice is characterized by compromised barrier function, and reduced mucus expression. 10 Studies have shown the presence of a leaky gut with impairment of intestinal barrier functions in genetically predisposed lupus-prone MRL/lpr mice. 11 Moreover translocation of E. gallinarum to liver and spleen under specific-free pathogen conditions was demonstrated to occur in autoimmune-prone (NZW x BXSB)F1 mouse, with induced expression of type I IFN and other proinflammatory cytokines in hepatocytes. 12 E. gallinarum reduced gut barrier-tightening molecules in (NZW x BXSB)F1 mice and in monocolonized non-autoimmune C57BL/6 mice. 12 Moreover, germfree or specific-germ free C57BL/6 mice treated with imiquimod have increased permeability to FITC-dextran with translocation of Lactobacillus reuteri to the mesenteric lymph nodes or the liver. 13 Gut permeability modification by resistant starch in this model was hypothesized by the authors to be related to tight-junctions alteration. 13 However other studies failed to demonstrate spontaneous leaky gut in lupus patients or mice.6,7
Calprotectin is a calcium-binding protein found in neutrophilic granulocytes; because it resists metabolic degradation and can be measured in feces with ease, fecal calprotectin is a reliable marker of inflammation or damage in the gastrointestinal tract. 14 Although fecal calprotectin has been classically associated with increased intestinal permeability and gut immune activation in inflammatory bowel diseases, 15 and serum calprotectin correlated to lupus activity, 16 its relation to intestinal permeability has been scantly investigated in lupus. 17
Accordingly, we aim to investigate fecal calprotectin kinetics, intestinal tight junction dysfunction and liver inflammation pathways in wild-type murine imiquimod- induced lupus for an improved understanding of gut-liver axis in lupus.
Methods
Animals and experimental protocol
The present study was approved by the Ethical Committee of the Saint Joseph University of Beirut. The protocols were designed according to the Guiding Principles in the Care and Use of Animals approved by the Council of the American Physiological Society and were in adherence to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication no. 85‐23, revised 1996), and according to the European Parliament Directive 2010/63 EU. The study was conducted on 8–10-week-old female C57BL/6J mice. Animals were housed at a stable temperature (25 °C) and humidity (50 ± 5%) and were exposed to a 12:12 h light–dark cycle. They were fed ordinary rodent chow, had free access to tap water, and were acclimatized for at least 1 week under these conditions before the start of the study. Mice were divided into two groups (n = 8 per group): imiquimod-treated group and Sham group. In the imiquimod-treated group, the skin on the right ears of each mice were treated topically, three times per week, with 1.25 mg of 5% imiquimod cream (Aldara 3 M Pharmaceutical, Saint Paul, MN, USA), this protocol was followed for six consecutive weeks.
Plasma biochemical assays
At sacrifice (day 45), mice were sedated by intraperitoneal injection containing a mixture of ketamine (Ilium, Australia; 75 mg/kg) and xylazine (Interchemie, Holland; 10 mg/kg). Pedal withdrawal reflex was performed to make sure of adequate depth of anesthesia. When animals were completely nonresponsive to toe pinching, blood samples were collected from the jugular vein in EDTA tubes. Whole blood was centrifuged at 4,500 RPM for 10 min. Plasma were aliquoted and stored at –80°C for plasma biochemical assays to avoid freeze-thaw cycles. The plasma concentrations of anti-dsDNA, C3, C4 and ACTα2 were quantified using enzyme-linked immunosorbent assay (ELISA) kits, following the instructions of the manufacturers. Anti-dsDNA, C3 and C4 ELISA kits (CSB-E12912m, CSB-E11194m, CSB-E08667m, CSB-E08707m respectively) were provided from Cusabio Technology LLC (Cusabio, Houston, TX 77,054 USA). No significant cross-reactivity or interference was observed in any kit.
Fecal calprotectin measurement
Fecal samples were collected at days 0, 22 and 44 in metabolic cages. Calprotectin levels were measured by ELISA kit according to the manufacturer protocol (Immundiagnostik AG, Bensheim, Germany).
Urine analyses
Urine samples were directly and aseptically withdrawn by acupuncturing the bladder at the time of sacrifice under anesthesia. Urine creatinine measurement was based on the Jaffe reaction of creatinine with alkaline picrate using the creatinine reagent (CP ABX Pentra), and absorbance was read at 510 nm (Horiba Medical, Montpellier, France). The ACR was calculated.
Histological assessment
At the time of sacrifice, the kidneys, liver and intestine were removed and perfused with ice‐cold Tyrode solution until all blood was removed. Tissues were kept at −80°C for protein extraction, whereas the other half fixed in 10% formalin solution (Sigma‐Aldrich, St. Louis, MO, USA). Neutral buffered formalin was used with a pH of 7.0 stabilized by the addition of sodium dihydrogen phosphate monohydrate (NaH2PO4.H2O) and disodium hydrogen phosphate anhydrous (Na2HPO4). The formalin‐fixed tissue was embedded in paraffin, and sections of 5 μm thickness were cut. Paraffin‐embedded sections of the tissues were stained with either hematoxyline-eosine (HE) or Masson trichrome (MT) for histopathological evaluation (Sigma‐Aldrich, St. Louis, MO, USA). Histological examinations were performed by two independent pathologists blinded to the conditions. A semi-quantitative scoring system was used.
For the intestinal tract, histological evaluation included the presence and intensity of active inflammatory infiltrate, scored 0 (absent), 1 (mild) and 2 (moderate) and the presence or absence of villous atrophy. For the liver steatosis was assessed, using a histological score ranging from 0 to 3 (score 0: <5%; score 1: 5–33%; score 2: 33–66%; score 3: >66%), Necroinflammation was scored 0 (absent), 1 (mild) or 2 (moderate) depending on the number of inflammatory infiltrates and apoptotic bodies. Mild necroinflammation referred to few lobular aggregates of inflammatory cells with or without apoptotic bodies. Necroinflammation was considered moderate when at least one lobular area contained two or more of such aggregates. Fibrosis was scored as absent (0), mild (1) or moderate (2) depending on the extent of extracellular matrix deposition. 18
Finally, for the kidney additional sections were stained with Jones methenamine silver (JMS). Glomerular lesions were graded semi-quantitatively on a scale of 0 to 2 or of 0 to 3 for endocapillary proliferation, mesangial matrix expansion, and segmental sclerosis (0 = <10%; 1 = 10–50%; 2 = >50% of the glomeruli examined), 2 for mesangioproliferation and for the presence of neutrophils infiltration (0 = <5%; 1 = 5–25%; 2 = 25–50%; 3 = >50% of the glomeruli). Global glomerular lesion scores were calculated for each mouse with at least 50 glomeruli. Total G score is the sum of mesangioproliferation, mesangial matrix expansion, neutrophil infiltration, endocapillary hypercellularity and segmental sclerosis scores.
Western blotting
Proteins were extracted from kidneys, liver and intestine tissues using RIPA buffer with protease and phosphatase inhibitors. Protein concentration was determined using the Bradford protein assay (Bio-Rad, Marnes-la-Coquette, France) using bovine serum albumin (BSA) as standard (Bio‐Rad, USA). Proteins were separated by SDS 10% PAGE and then blotted on polyvinylidene fluoride (PVDF) membrane (Bio‐Rad Laboratories Inc., Irvine, CA, USA). Membranes were blocked in TBS‐Tween blocking solution (in mmol/l: Tris‐HCl 100, NaCl 150, and Tween‐20 0.1%) with 5% non-fat milk or 5% BSA and incubated with the various antibodies: Tight junction protein (ZO1) (1/1500; ab96587; Abcam, Cambridge, UK); Claudine (1/500; ab19098; Abcam, Cambridge, UK); Occludine (1/50000; ab167161; Abcam, Cambridge, UK); Toll Like Receptor 4 (TLR4) (1/500; ab22048; Abcam, Cambridge, UK); TLR7 (1/100; ab24184; Abcam, Cambridge, UK); Nuclear Factor kappa-light-chain-enhancer of activated B cells (NF-kB) (1/2000; ab16502; Abcam, Cambridge, UK); phosphorylated NFkB (pNFkB) (1/2000; ab86299; Abcam, Cambridge, UK); glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (1/2500; D6H11; Cell Signaling Technology; MA, USA); T-cell immunoglobulin or Kidney Injury Molecule -1 (KIM-1) (1/500; ab47634; Abcam, Cambridge, UK); Neutrophil gelatinase-associated lipocalin (NGAL) (1/500; ab63929; Abcam, Cambridge, UK); Escherichia Coli (1/4000; ab25823; Abcam, Cambridge, UK). Visualization was done using enhanced chemiluminescence and signals detected by an imaging system equipped with a CCD camera (Omega Lum G, Aplegen, Gel Company, SF, USA). Quantifications were performed using ImageJ software. Three western blots were performed for each condition.
Statistical analysis
All quantitative data are reported as mean ± SEM. Statistics were analyzed using SigmaPlot Software (version 12.5). The normal distribution of the values was checked by the Shapiro–Wilk test. When the normal distribution was met, one‐way analysis of variance (ANOVA) tests were performed for multiple comparisons of values and post hoc Holm–Sidak tests were performed to identify which group differences accounted for significant overall ANOVA results. When normal distribution was not met, the Kruskal–Wallis one‐way ANOVA on ranks was performed followed by the Mann–Whitney U tests. All values with p < 0.05 were considered significant.
Results
Plasma autoantibodies and complements
Anti dsDNA plasma concentration were positive in imiquimod group than in control (21.77 ± 1.91 ng/mL versus 13.08 ± 0.52 ng/mL, respectively; p < 0.041). Moreover, C3 and C4 plasma concentration were significantly lower in imiquimod group when compared to control one (C3: 1297 ± 20 μg/mL versus 2079 ± 250 μg/mL; C4: 24.62 ± 12.75 μg/mL versus 89.82 ± 17.71 μg/mL, respectively; p < 0.029).
Lupus nephritis assessment
Significant increase in mesangial proliferation, endocapillary proliferation, segmental glomerulosclerosis, neutrophil infiltration, and inflammation scores were observed in imiquimod group (Figure 1). Moreover, protein expressions of KIM-1 and NGAL were significantly increased in imiquimod group with a trend toward increased urine ACR (Figure 2).
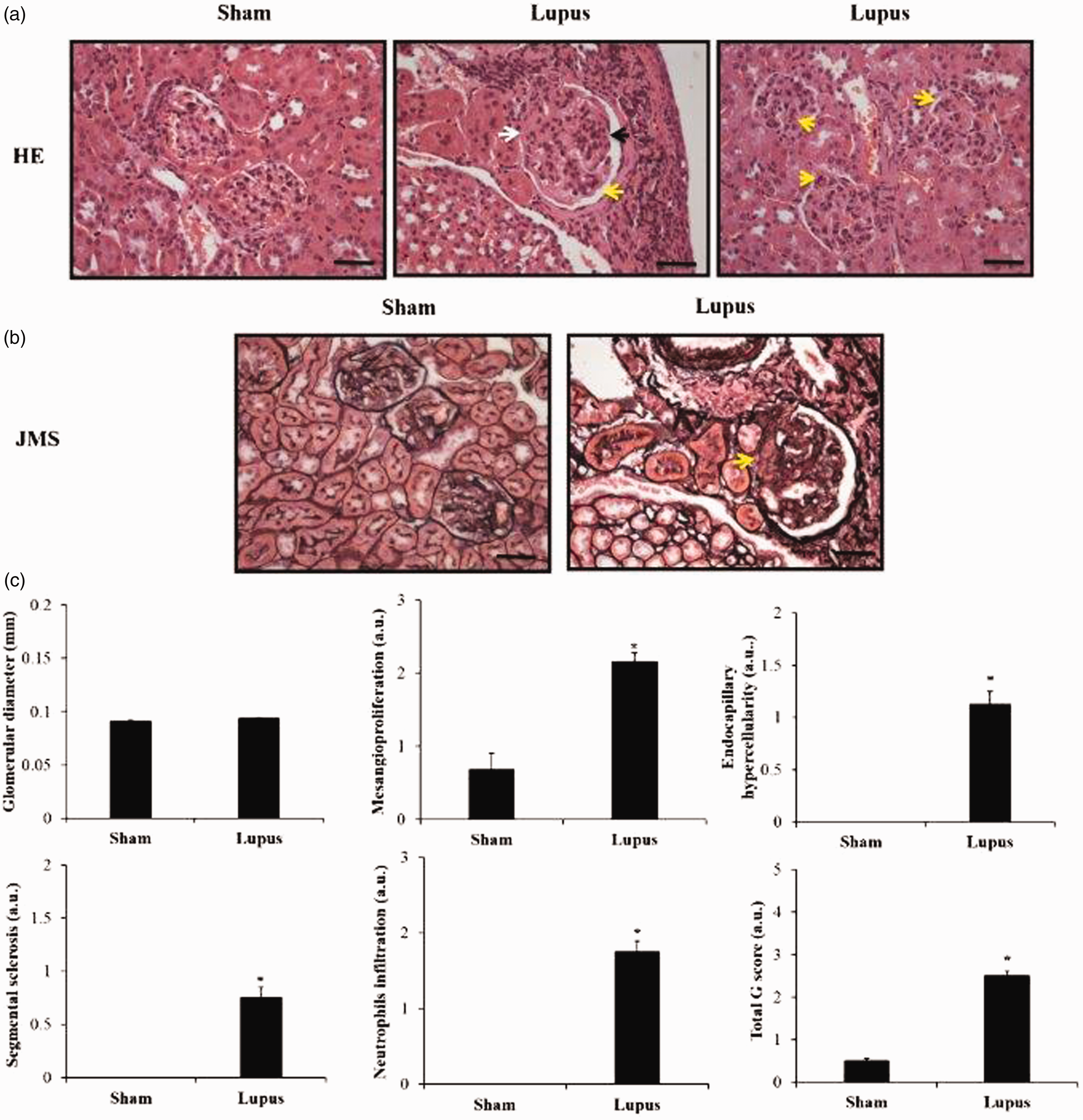
Microscopic assessment of renal sections and histopathologic scores. (a) HE-stained renal section from Sham and Lupus mice showing mesangial proliferation and mesangial matrix expansion (black arrow), segmental sclerotic lesion (white arrow), and endocapillary hypercellularity with neutrophils infiltration (yellow arrows). (b) JMS-stained renal section from Sham and Lupus mice showing the segmental sclerotic lesion (white arrow). Scale bar = 40 µm; x40 magnificence. (c) Histograms showing glomerular diameter, mesangial proliferation, endocapillary hypercellularity, segmental glomerulosclerosis, neutrophil infiltration, and inflammation scores. Total G score is the sum of mesangioproliferation, mesangial matrix expansion, neutrophil infiltration, endocapillary hypercellularity and segmental sclerosis scores. a.u.: arbitrary unit. * p < 0.05 vs Sham.
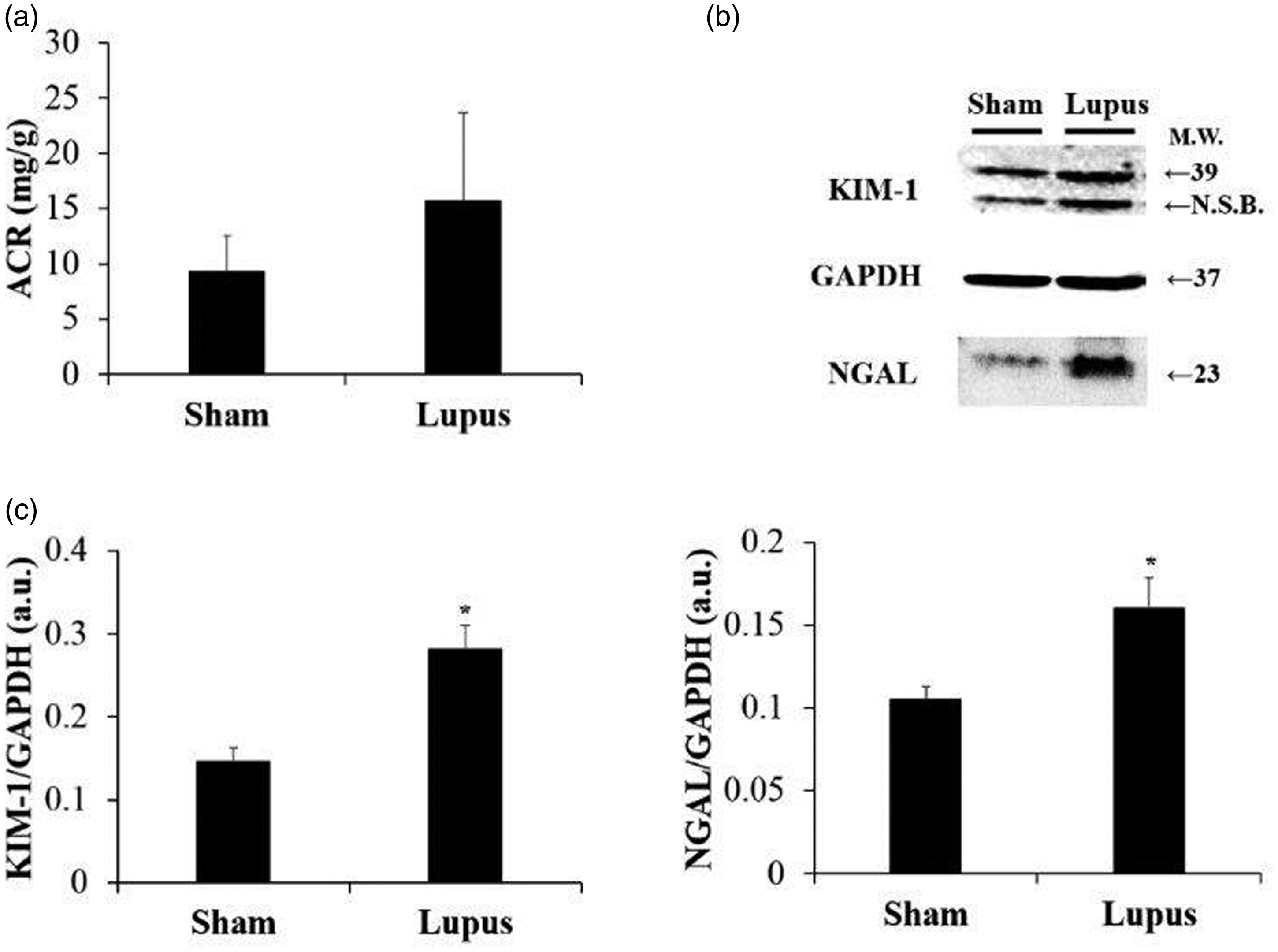
kidney injury markers. (a) ACR ratio in urine. (b) Representative Western blot images of KIM-1 and NGAL. (c) Histograms showing KIM-1 and NGAL protein expressions in renal tissue, normalized to GAPDH, in Sham (n = 8) and Lupus (n = 8) groups. a.u.: arbitrary unit. *p < 0.05 vs Sham.
Intestinal histopathology, fecal calprotectin and tight junction assessment
No inflammatory infiltrates, fibrosis, or villous atrophy were found in imiquimod and control groups (Figure 3(a)). Besides no significant difference was found in baseline fecal calprotectin between groups. However, at days 22 and 44, fecal calprotectin was significantly more elevated in imiquimod-group compared to control (Figure 3(b)). Finally, protein expressions of Claudine, Zonula Occludens-1 (ZO-1) and occludin were significantly decreased in imiquimod group when compared to control (Figure 4).
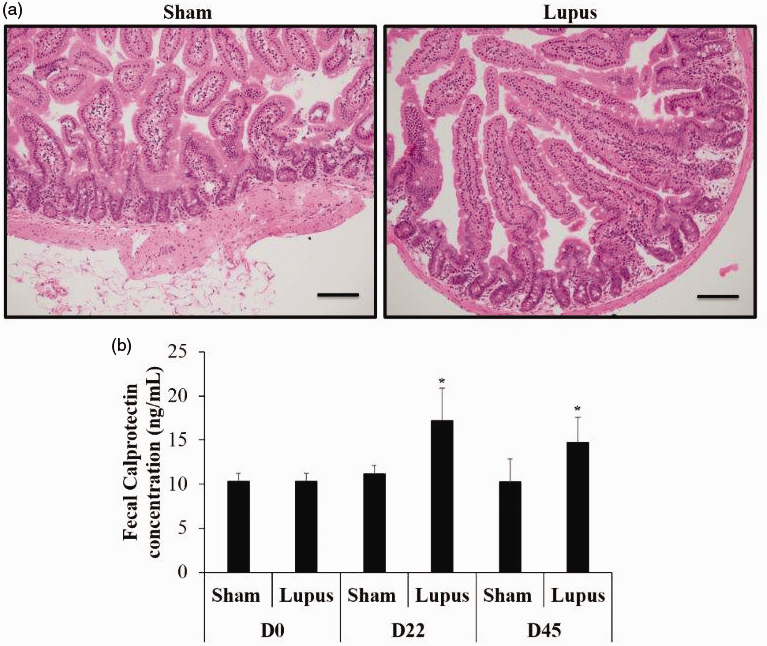
Intestinal histopathology and fecal calprotectin. (a) Normal intestinal histology in Sham and Lupus groups. (b) Histogram showing fecal calprotectin at day 0, 22 and 44 in Sham (n = 8) and Lupus (n = 8) groups. *p < 0.05 vs Sham.
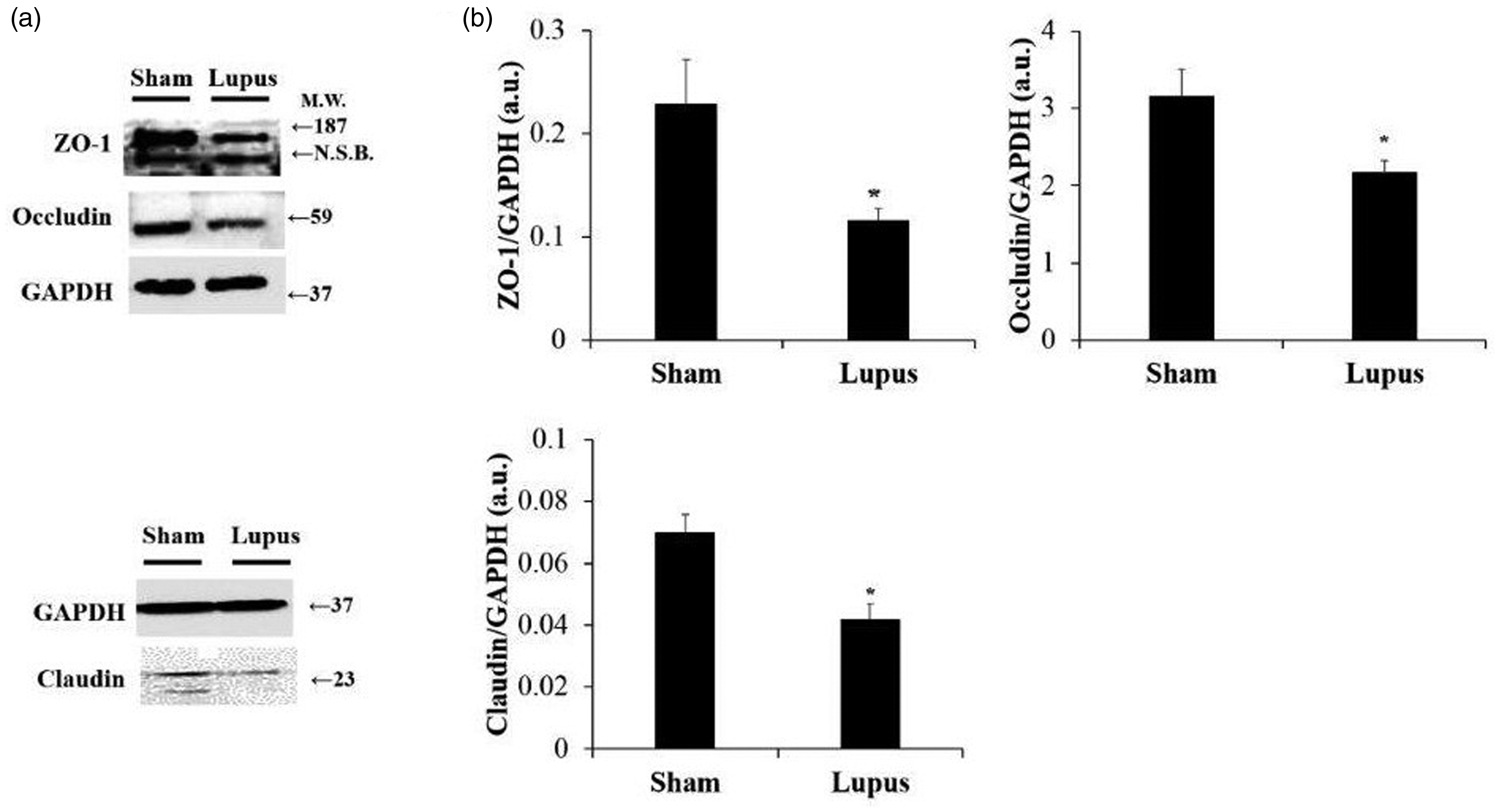
Intestinal tight junction protein assessment. (a) Representative Western blot images of ZO-1, occludin and claudin. (b) Histograms showing ZO-1, occludin and claudin protein expressions in intestine tissue, normalized to GAPDH, in Sham (n = 8) and Lupus (n = 8) groups. a.u.: arbitrary unit. * p < 0.05 vs Sham.
Liver histopathology and inflammatory markers
No fibrosis was observed in both groups. However, steatosis and necro-inflammation scores were significantly higher in imiquimod group (Figure 5(a) and (b)). Moreover, protein expression of E. coli was highly elevated in imiquimod group when compared to control (p < 0.05) (Figure 5(c) and (d)). Additionally, liver protein expression of TLR4, TLR7, NFκB, pNFκB, and pNFκB/NFκB were significantly higher in imiquimod group (Figure 6).
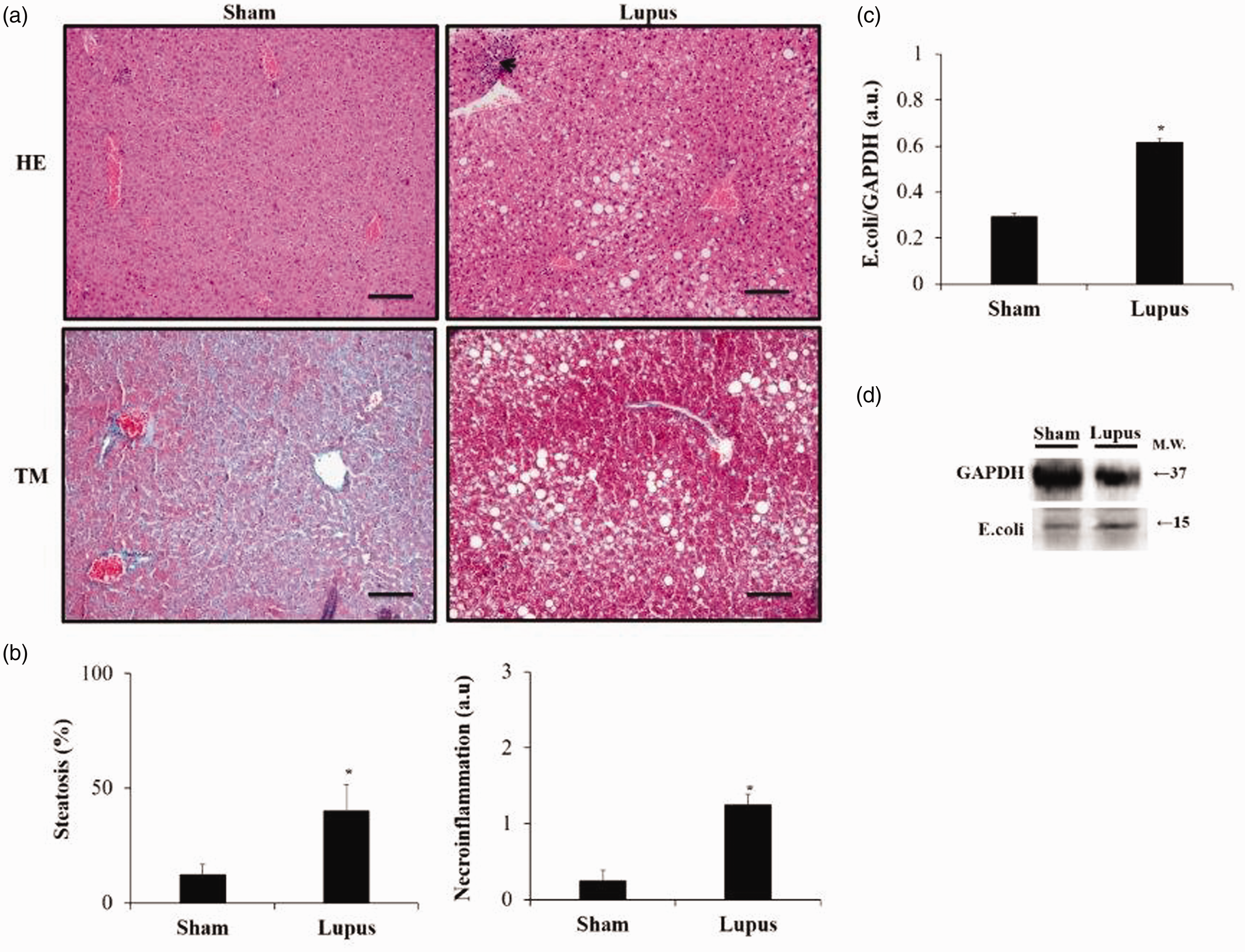
Liver Histopathology. (a) Microscopic assessment of liver sections stained with HE and TM. (b) Histograms showing steatosis and necro-inflammation (arrow). Scale bar = 100 µm. (x20) magnificence. (c) Representative Western blot image of E.coli. (d) Histograms showing E.coli protein expressions in liver tissue, normalized to GAPDH, in Sham (n = 8) and Lupus (n = 8) groups. a.u.: arbitrary unit. * p < 0.05 vs Sham.
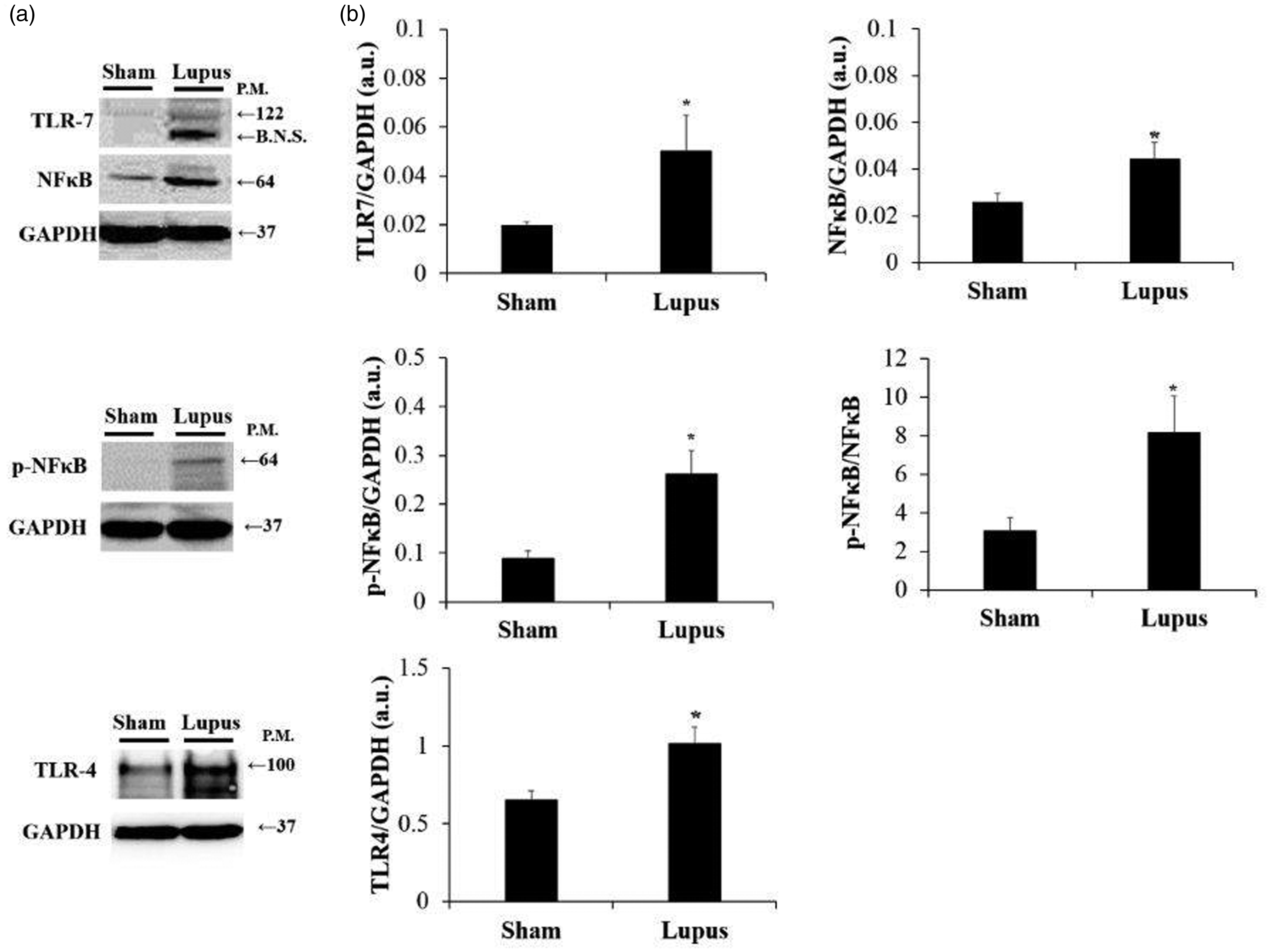
Liver protein expression of TLR-NFκB. (a) Representative Western blot images of TLR4, TLR7, NFκB, and pNFκB. (b) Histograms showing protein expressions of TLR4, TLR7, NFκB and pNFκB in liver tissue, normalized to GAPDH, in Sham (n = 8) and Lupus (n = 8) groups; and pNFκB/NFκB ratio. TLR4: TLR7; NFκB: pNFκB: a.u: arbitrary unit. *p < 0.05 vs Sham.
Discussion
To our knowledge, this study is the first to follow elevation of fecal calprotectin and to explore liver TLR4- NFκB pathway in a model of induced lupus in wild-type mice, highlighting the role of gut permeability as a bridge between environmental activators of innate immunity and autoimmunity induction in nonautoimmune strains.
Our study focuses on the gut-liver axis role in lupus pathogenesis in a wild strain exposed to epicutaneous TLR7 agonist. While spontaneous murine lupus models have proven valuable for dissecting pathogenic mechanisms and ultimately for clarifying the genetic basis for systemic autoimmunity, induced lupus models in nonautoimmune strain shed light on potential mechanisms by which environmental exposure can contribute to the development or severity of lupus. 19
Gut permeability dysfunction, as systemic manifestations in lupus, could be driven by TLR-7. In lupus patients, abnormal stimulation of TLR7 and TLR9 by self-nucleic acids was demonstrated to contribute to persistent production of type I IFN, a key player in the pathogenesis of lupus. 2 In addition, TLR7-mediated IFN-α production is up-regulated in peripheral blood mononuclear cells from lupus patients and correlates positively with lupus activity. 19 Furthermore, TLR7 seems to be implicated in leaky gut pathogenesis in autoimmune-prone (NZW x BXSB)F1 mouse, which carries a TLR7 gene duplication. 12 However, in other models of TLR7-induced auto-immune diseases, gut permeability dysfunction was not observed: for example, in one model of imiquimod induced dermatitis (62.5 mg applied topically on the backs of B6 mice for six consecutive days, followed by three days for rest, for two cycles), intestinal permeability was not decreased at two weeks as measured by plasma levels of FITC-dextran and tight junctions assessment (ZO-1, occludin, and claudin-2). 20
TLR-7 activation through topical TLR-7 agonist, imiquimod, constitutes the base of the model of inducible systemic lupus erythematosus used in our study. The present study replicates the finding that wild-type mice exposed to epicutaneous application of 1.25 mg of 5% imiquimod, three times weekly, develop lupus-like systemic auto-immune disease. 2
Positive anti-DNA antibodies with increasing urine protein to creatinine ratio were documented in our results confirming the biological activity of lupus. This was associated to histopathological lesions of glomerulonephritis and elevated of kidneys injury markers KIM-1 and NGAL. KIM-1 is a transmembrane protein in renal proximal tubule cells and is believed to play a role in tubulo-interstitial damage. 20 The potential use of urinary KIM-1 levels to screen for active lupus nephritis and for predicting renal damage such as glomerular nephritis, tubulo-interstitial inflammation, and tubular atrophy was demonstrated in several studies. 21 NGAL is a protein of the lipocalin family that is highly expressed in activated leukocytes and other types of cells, including tubular epithelial cells with injury. 22 Kidney binding of pathogenic antibodies (like anti-DNA) stimulates local expression of NGAL, which plays a crucial role in the pathogenesis of nephritis via promotion of inflammation and apoptosis. 23 Herein, KIM-1 and NGAL levels were clearly associated with lupus nephritis. 21
In addition to common organ damage classically associated with lupus, recent studies have pointed to a dysfunction of the intestinal permeability in lupus-prone mice and germ-free wild-type mice10–13 but these observations were inconsistent across literature . Herein we are the first to give evidence for a loss of tight junction proteins in TLR7-agonist induced lupus in wild-type mice raised in standard conditions and the first to show a significant progressive increase of fecal calprotectin. In comparison to other organ lesions, gut permeability dysfunction in our study seems to precede the installation of renal injury documented to start at four weeks in the original description, 2 but seems to be concomitant to the development of splenomegaly, vasculopathy and rising auto-antibodies demonstrated to occur at three weeks in a recent study on a similar model. 3
In our study, significant elevation of fecal calprotectin was observed at day 22 of imiquimod treatment and persists till sacrifice, suggesting activation of inflammatory cascades in the intestinal mucosa and disruption of gut permeability. To the best of our knowledge, our study is the first to assess the timeline course of fecal calprotectin in lupus. Calprotectin is a member of the S100 family of zinc- and calcium-binding proteins and is a heterodimer of S100A8/9. It accounts for approximately 60% of cytosolic protein in neutrophil. 14 Any disruption to the mucosal architecture of the gastrointestinal tract allows neutrophils that accumulate at sites of active inflammation to be released into the lumen and subsequently shed in feces. 24 The fecal concentration of calprotectin has been shown to strongly correlate with the gold standard indium-111–labeled granulocyte test and is directly related to the extent of inflammation in inflammatory bowel diseases. 14 Besides, fecal calprotectin have been related to exercise-induced gut barrier dysfunction in healthy individuals, 25 with no documented macroscopic intestinal lesions. Fecal calprotectin modification is also observed in healthy Wistar Furth rats after diet modification without macroscopic intestinal inflammation. 26 Thus, the elevation of fecal calprotectin could be a marker of gut inflammation and subsequent neutrophil recruitment, or to increased gut permeability to neutrophils. 26
Protein expression of Claudine, ZO-1 and Occludin in intestinal tissue was significantly decreased in imiquimod group. Moreover, tight junction loss was accompagnied by the presence of E. coli protein expression in the liver tissue; this is consistent with a recent study on experimental auto-immune hepatitis. 27 Tight junctions are generated by the assembly of multiple proteins located near the apical part of the epithelium between neighboring cells and control the permeability of the paracellular transport pathway. They consist of two functional protein categories, integral transmembrane proteins (like occluding and claudins) that form a network between adjacent cell membranes, and peripheral membrane or plaque proteins (like ZO-1) acting as bridges to connect integral membrane proteins to the actin cytoskeleton and to other signaling proteins. 28 Proinflammatory cytokines such as TNF-α, IL-1β, and IFNγ promote tight junction permeability disruption. 29 Stimulation by INF- rapidly enhanced disruption of the intestinal barrier and permeability in a transwell in vitro model of intestinal endothelial barrier. This is mediated by mislocalization of ZO-1, claudin-5, and occludin accompanied by dramatic depolymerization of the peri-junctional actin cytoskeleton. 30 Furthermore, INFγ causes a dramatic, dose-dependent increase in E. coli translocation, across human colonic epithelial monolayers. 31
Dysbiosis and leaky gut are mainly documented in spontaneous animal models of lupus. The composition of the gut microbiota changes markedly before and after the onset of lupus disease in NZB/W F1 mice.32,33 In the MRL/lpr model of lupus nephritis, a marked depletion of gut Lactobacillales was found, and a “leaky gut” that could be reversed by Lactobacillus colonization.11,33 Furthermore, in MRL/lpr mice, antibiotics given after disease onset ameliorated kidney histopathology and increased the intestinal barrier function. 34 In SNF1 lupus prone mice, abundant Rikenellaceae was associated with more rapid lupus progression. 35 Recent data point to a clear causative potential of Enterococcus gallinarum with anti-dsDNA antibody production after translocation to the liver in (NZW x BXSB)F1 mice. 12 As for inducible models of lupus, leaky gut was solely studied in germ-free or monocolonised C57BL/6 mice after TLR7 agonist administration in a few studies.12,13 In our model, decreased intestinal permeability was documented after induction with imiquimod in nonautoimmune strain without intentional modification of the physiologic intestinal microbiome. A future study is planned by our team to analyze microbiome alteration under imiquimod treatment.
In addition to experimental models, human studies add to the complexity of host-microbiome interactions in lupus and suggest that influences of lupus disease activity and gut dysbiosis may be bidirectional. 36 Specific strains of a gut commensal (like Ruminococcus gnavus of the Lachnospiraceae family), may contribute to the immune pathogenesis of lupus 17 ; alternatively, these shifts may be secondary, as immune activation and systemic inflammatory milieu could alter the metabolomic environment in the gut that drives dysbiotic shifts. 36 Decreased ratio of Firmicutes to Bacteroidetes compared to healthy controls is observed in lupus patients37–39 while other studies demonstrated relative abundance of Lactobacillus species in lupus patients. 13 Gut commensals of lupus patients share protein epitopes with the Ro60 autoantigen and Ruminococcus gnavus strain cross-reacts with native DNA.17,40,41 Parents and children of lupus patients exhibit elevated plasma LPS levels compared to controls with positive correlation between LPS levels and anti-double-stranded DNA IgG levels. 42
Once intestinal tight junctions are disrupted, the intestinal mucosal permeability increases, which elevates LPS contents in the liver. 43 When LPS enters the liver, it synergizes with TLR4, a congenital immune receptor expressed in hepatocytes, and ultimately causes NFκB translocation. TLR4-NFκB is a highly conserved and tightly regulated signaling pathway central to liver survival and homeostasis. NF-κB regulates the expression of inflammatory factors and induces liver inflammation. 44 Overall, the LPS-TLR4-NFκB pathway is a bridge between the intestinal homeostasis and liver inflammation. Our results confirm liver inflammation observed in the original model 2 and adds new data on innate immunity markers in the liver with increased hepatic protein expression of TLR4, TLR7, NFκB, pNFκB, and pNFκB/NFκB and E.coli translocation.
In conclusion, our study contributes to the understanding of gut-liver axis in lupus pathogenesis and manifestations and demonstrates an early rise of fecal calprotectin in imiquimod-induced lupus in wild-type mice. Tight junctions loss could be of particular interest as a potential target of future therapeutic interventions on gut-liver axis and fecal calprotectin a useful early surrogate marker of gut permeability loss in lupus.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the research council of the Saint Joseph University of Beirut – Faculty of Medicine, project FM382.
Contributorship
GM and NF designed the research, performed experiments, analyzed the data and drafted the manuscript. JH, CN and MC performed experiments and helped for data acquisition and analysis. HN and VS helped for histology interpretation. YS helped to review the manuscript. All authors discussed the results and approved the final version of the manuscript.