
Abstract
Antiphospholipid syndrome (APS) is a chronic systemic autoimmune disease characterized by venous, arterial, and microvascular thromboses and/or recurrent pregnancy morbidity, that occur in the persistent presence of antiphospholipid antibodies (aPL). APS can present with a wide range of clinical manifestations often reffered as “extra-criteria”. These features, although apparently less common, can severely impact patients’ outcome. Here, we report the case of a patient with a newly diagnosed APS. He previously experienced a recurrence of venous thrombosis after discontinuation of anticoagulant therapy in association with cutaneous ulcerations as presenting symptoms. Interestingly, skin lesions did not improve with full anticoagulant treatment. Due to concomitant presence of thrombotic and microvascular involvement, immunomodulatory therapy with steroid pulses followed by intravenous injections of belimumab was started, with progressive and significant amelioration, leading to complete recovery. Following the presentation of the current case report, we highlight the importance of suspecting APS in young patients experiencing unprovoked thrombosis. We also emphasized the critical issue of testing aPL during anticoagulant treatment and focused on the need of aPL retesting in patients with positivity at high titers. We also highlight the double nature of aPL-mediated clinical manifestations. While most patients presented with pure thrombotic complications, one should always remember that APS is an autoimmune-mediated disease, which can benefit from alternative therapeutic approaches beyond anticoagulation.
Introduction
Antiphospholipid syndrome (APS) is a chronic systemic autoimmune disease characterized by venous, arterial, microvascular thrombosis, and/or recurrent pregnancy morbidity that occur in the persistent presence of antiphospholipid antibodies (aPL). The aPL included in the current classification criteria are lupus anticoagulant (LA), IgG and IgM anti-cardiolipin (aCL), and antiβ2-glycoprotein 1 (aβ2GP1) antibodies. These tests must be confirmed after at least 12 weeks. 1
APS can present with a wide heterogeneity of clinical manifestations beyond thrombotic events and pregnancy complications associated with the presence of aPL. These clinical features, usually referred as “extra-criteria” manifestations, include neurological disorders such as chorea and myelitis, hematological diseases, livedo reticularis, and aPL-nephropathy. 2 Recently, new classification criteria of the syndrome have been published, highlighting the importance of these non-thrombotic aPL-related clinical features. 1
At present, anticoagulant therapy with vitamin K antagonists (VKA) with an international normalized ratio (INR) of 2–3 is the mainstay of treatment for APS and history of venous thromboembolism (VTE). Despite adequate time in therapeutic range (TTR), the patients could develop both arterial and venous recurrences. Combining low-dose aspirin (LDA) with VKA or intensifying the anticoagulation to an INR target to 3–4 may decrease the risk of arterial events; however, there is no unanimous consensus among different treatment strategies. 3
Herein, we present the case of a patient with APS, who had a recurrence of venous thrombosis and skin lesions at the right foot toes as presenting symptoms. We aim to highlight that APS is a heterogeneous disease with a broad spectrum of clinical features and the crucial role of making timely diagnosis and choosing the best therapeutic approach.
Case presentation
In January 2023, a 53-year-old male patient presented at our department for a suspected newly diagnosed APS.
The patient had no previous personal and family history of known autoimmune disease and at the time of the evaluation, among conventional cardiovascular risk factors, only arterial hypertension on pharmacological therapy was recorded and adequately controlled with doxazosin 0.5 mg and carvedilol 6.25 mg daily.
In November 2021, he experienced an idiopathic and unprovoked deep vein thrombosis (DVT) of the left popliteal vein, confirmed by color Doppler ultrasound, treated for six months with rivaroxaban at the dose of 20 mg daily.
The patient was regularly followed up elsewhere, and in May 2022 the anticoagulant therapy was suspended without performing additional diagnostic investigations. A few weeks after the interruption of anticoagulation the patient experienced a recurrence of DVT at the same site, assessed by color Doppler ultrasound. At that time, a prolonged partial thromboplastin time (aPTT) was found and therefore the patient underwent further testing, including complete aPL profile, showing LA positivity (aPTT screen 3.14, mix 2.44, confirm 1.26, dRVVT screen 2.16, mix 1.62, and confirm 2.06), along with aβ2GP1 IgG (317 U/mL) positivity and negative aCL IgG and IgM and aβ2GP1 IgM.
Considering the strong suspicion of APS, the patient has resumed anticoagulant treatment with VKA at INR target 2–3. After 12 weeks, LA testing was confirmed as positive (aPTT screen 3.74, mix 2.67, and confirm 1.1; dRVVT screen 2.59, mix 1.85, and confirm 1.05), but aβ2GP1 and aCL IgG and IgM autoantibody testing was not performed. The patient was also found to be positive for anti-nuclear antibodies (ANA) 1:160 titer fine speckled. Levels of erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), full blood count, complement levels both for C3 and C4, extractable nuclear antigens (ENA), and anti-double-stranded DNA (anti-dsDNA) antibodies were negative or within normal range. No cause of inherent thrombophilia was detected.
In January 2023, the patient presented with painful and violaceous skin lesions at the right foot toes. At that time, INR was in range (INR target 2–3) based on available records. A diagnosis of calciphylaxis was hypothesized by the general practitioner, with VKA as the precipitant factor. VKA was therefore stopped and replaced with fondaparinux 7.5 mg once daily. No clinical improvement was observed over the following 3 weeks. Chest radiography and abdomen ultrasonography were performed with no evidence of pathological lesions. Blood tests including kidney function and serum calcium levels were within the normal range.
When the patient first came to our attention (end of January 2023), skin lesions were still present (Figure 1). At the time of our evaluation, the patient presented with normal blood pressure under anti-hypertensive treatment as previously described, while no other traditional cardiovascular risk factor was detected (including smoking, diabetes, and dyslipidemia). Moreover, no additional clinical feature compatible with connective tissue disorder was present (including, among others, Raynaud’s phenomenon, aphthosis, xerostomia, xerophthalmia, photosensitivity, and defluvium). First, the patient underwent a color Doppler ultrasound that resulted negative for stenosis, vascular occlusions, or for the presence of atherosclerotic plaques. After ruling out the hypothesis of VKA-associated calciphylaxis, based on the absence of renal dysfunction and normal serum calcium levels and the other possible causes, such as infections and paraneoplastic syndrome, an association between APS and microvascular involvement was hypothesized. Skin lesions at the right foot caused by APS-related microvascular damage.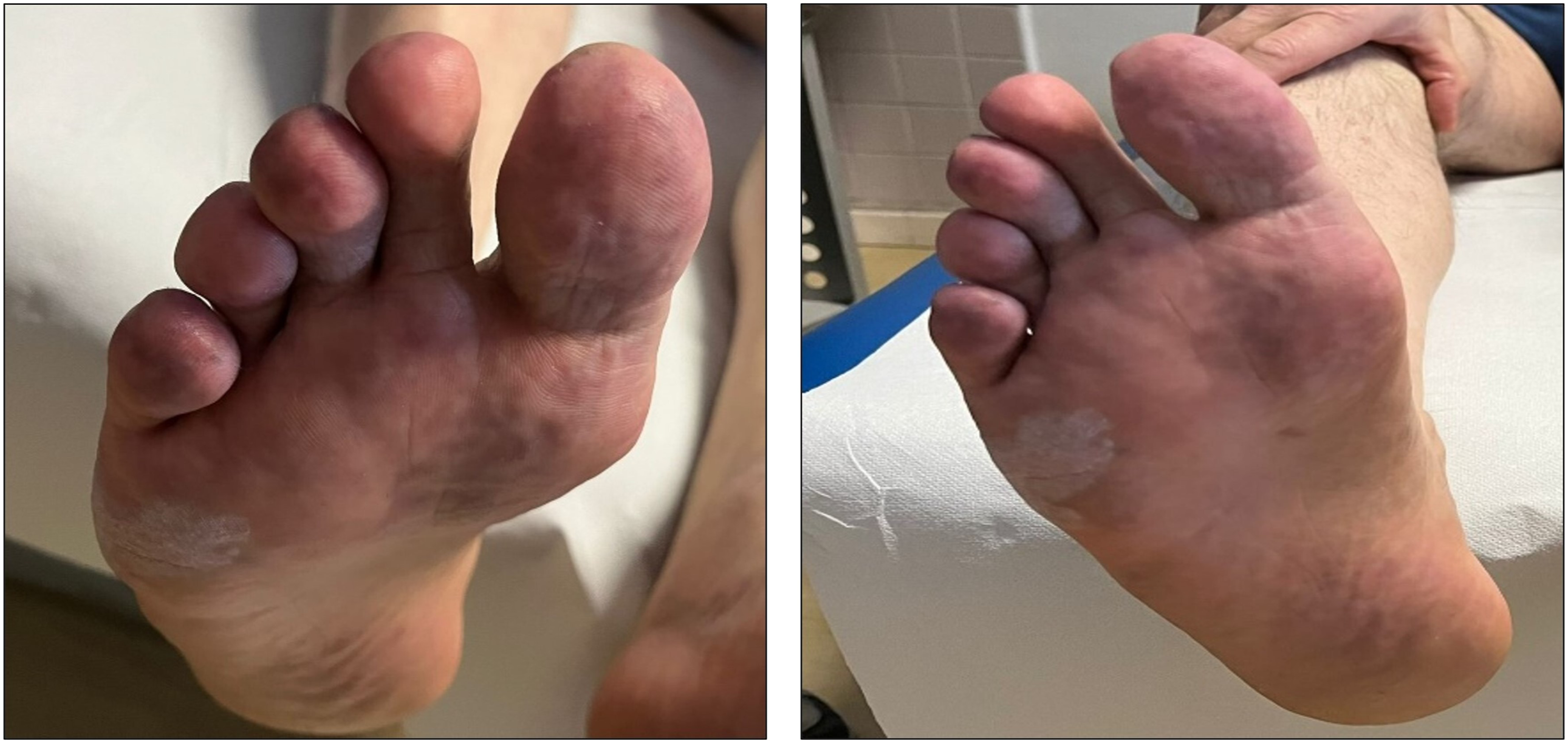
The patient underwent treatment with vasodilator agent (intravenous synthetic prostanoids), LDA, and steroids (prednisone at the initial dose of 25 mg daily, rapidly tapered and suspended within 2 weeks) combined with fondaparinux 7.5 mg once daily as anticoagulation. aPL testing was repeated showing a triple-positive profile with LA, aβ2GP1 IgG, and aCL IgG positivity. ANA, ENA, and anti-dsDNA were found negative.
It has to be acknowledged that we did not perform a skin biopsy or positron emission tomography, in order to rule out vasculitis, based on the absence of signs and symptoms compatible with a systemic inflammatory process, such as the bilateral involvement of lower limbs, increased levels of acute-phase proteins (including ESR, CRP, complement fractions, albumin, and ferritin), as well as evocative findings at the color Doppler ultrasound of the right leg and foot.
Taking into account the early recurrence of venous thrombosis and the presence of APS-related microvascular disease despite anticoagulant therapy, in February 2023 the patient was enrolled in “The Belimumab Antiphospholipid Syndrome Trial” (BLAST), an open-label, prospective, phase II descriptive pilot trial for refractory and/or non-criteria manifestations of APS, and he received the immunomodulatory therapy with Belimumab (a human immunoglobulin-G1λ monoclonal antibody that inhibits the biologic activity of soluble B-lymphocyte stimulator) 4 at the dose of 10 mg/kg monthly combined with VKA with INR target of 2–3 as anticoagulation.
At 1 month of follow-up the skin lesions were completely regressed, with no residual damage, excellent pain control, and full clinical recovery. The clinical course of the patient is summarized in Figure 2. Timeline of the clinical events experienced and the therapies received by the patients. DVT means deep vein thrombosis; aPL, antiphospholipid antibodies; LA, lupus anticoagulant; aβ2GP1, antiβ2-glycoprotein antibodies; aCL, anti-cardiolipin antibodies; VKA, vitamin K antagonists; INR, international normalized ratio; LDA, low-dose aspirin. 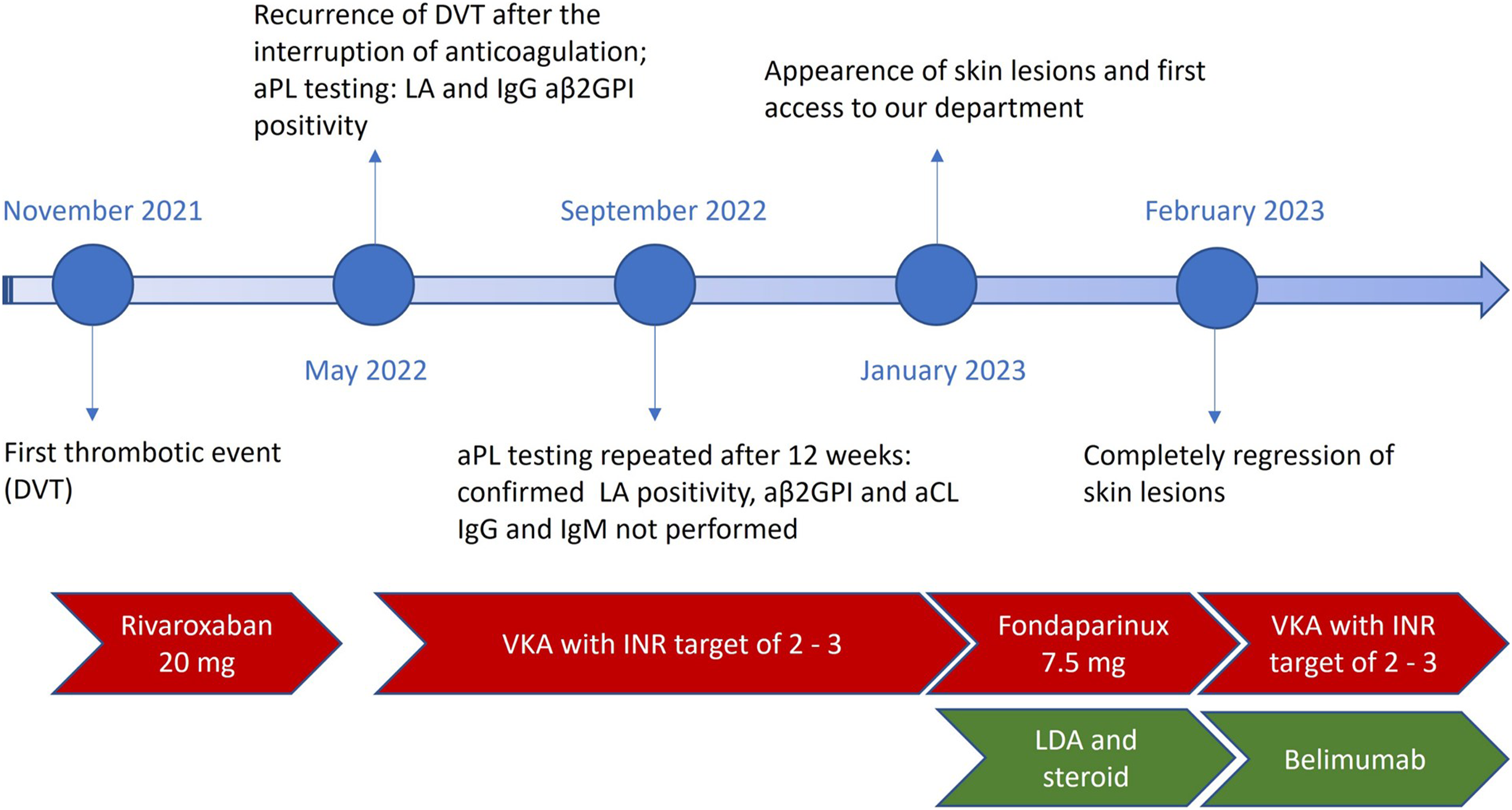
Discussion and conclusion
The present clinical scenario gives us the opportunity to address some clinical needs representing a challenge in the APS field, particularly on the management of patients with newly diagnosed or suspected APS.
Firstly, when do we need to consider aPL testing? Young patients with unprovoked thrombosis or provoked by weak factors, major thrombotic events, and atypical sites of vascular occlusion should be tested for the presence of aPL. 3 Making a diagnosis of APS might be easy, if you think about it. An early diagnosis is essential to choose the most appropriate anti-thrombotic therapy and reduce mortality rate, long-term disability, and damage.
In particular, while direct oral anticoagulants (DOACs) are an attractive alternative to VKA for the treatment of VTE, they are not recommended in patients with a defined diagnosis of APS, especially those with high risk aPL profile, such as persistent LA positive and triple aPL positive subjects. In fact, previous evidence has shown that rivaroxaban is inferior in terms of safety and effectiveness in high risk APS patients, when compared to VKA. Nowadays, VKA with a target INR range of 2–3 remains the mainstay for the treatment of acute VTE and for the secondary prevention of thrombosis. 3
On the other hand, anticoagulant therapy could negatively affect functional aPL testing. All anticoagulants prolong test clotting times and interfere with the assays used for LA detection, leading to both false-positive and false-negative results. Thus, do we need to stop anticoagulation for aPL testing? There is no consensus on the types of procedures to be adopted. Ideally, LA testing should be performed right after the first clinical manifestation of the disease, as already highlighted above, as part of the panel of routine investigations, before starting anticoagulant therapy. When not performed, DOACs should be stopped at least 48 hours before LA testing and patients on VKAs should be switched to low-molecular weight heparin. However, since discontinuation of anticoagulation might be problematic in clinical practice, mainly due to the increased risk of early recurrence, the employment of DOAC-removal agents constitutes a promising solution. 5
It is also important to mention that the timing of aPL testing is crucial to avoid misinterpretation, especially for LA determination. In fact, increased levels of coagulation factors such as factor VIII, which are observed during an acute thrombotic event, affect functional coagulation tests such as LA. In this view, current guidelines contraindicate performing aPL testing, especially LA detection, during thrombosis.
An alternative approach is represented by the introduction of the so called “extra-criteria aPL” as additional biomarkers for LA testing. Among numerous autoantibody specificities, IgG and IgM anti-phosphatidylserine–prothrombin complex (aPS/PT) has been shown to improve the accuracy of APS diagnosis. 6 While these tests are not meant to replace LA, they do not suffer for some of the limitations of functional assays and should be considered when LA result is not conclusive, not available or fluctuating.
The global anti-phospholipid syndrome score (GAPSS) has been developed as an additional tool to stratify the risk of thrombotic recurrences, considering traditional cardiovascular risk factors, such as dyslipidemia and arterial hypertension, as well as aPL profile including aPS/PT IgG and/or IgM positivity. The score has been originally developed for aPL-positive patients with a concomitant diagnosis of systemic lupus erythematosus, and subsequently applied in primary APS setting. 7 However, due to the limited availability of extra-criteria aPL testing, especially in routine laboratories, the GAPSS score can be replaced by a modified version, the adjusted GAPSS (aGAPSS).
In our case, at the time of the first clinical evaluation, aGAPSS was calculated with a result of nine out of 17, confirming the high risk of thrombotic recurrence. In fact, it is demonstrated that aGAPSS is significantly higher in patients with recurrent arterial or venous thrombotic events, than to those without recurrence. 8
Finally, considering the difficulties encountered in aPL testing while on anticoagulant therapy, do we need to repeat aPL after 12 weeks even in case of high positivity? In accordance with the new APS classification criteria 1 and the recommendations published by the Scientific Standardization Subcommittee (SSC) of the ISTH, the laboratory diagnosis of APS requires that aPL positivity is confirmed after at least 12 weeks, to avoid the risk of over-diagnosis and consequently of over-treatment. Indeed transient aPL positivity, especially at low titers, is known for being common as consequence of infections or some medications, with unclear clinical value. 9 However, a study performed by the APS Action group demonstrated that approximately 80% of clinically significant aPL profiles (especially triple positive rather than isolated LA) confirmed their persistence over time. 10 Furthermore, it has been shown that even double aPL positivity (e.g., positive for aCL and aβ2GP1, both IgG isotypes) is persistent at retest with higher frequency than isolated aPL profiles. It is also known that antibody titers are the strongest predictor for a persistent positivity: high-titers aPL remain stable, instead of low-titers aPL that can frequently fluctuate over time. Thus, these data suggest that retest may not postpone the use of VKA in patients with high-titer aPL positivity, allowing a more timely management of the patients.
This case remembered us that APS is a heterogenous autoimmune disease with a wide clinical spectrum that goes beyond thrombosis such as neurologic and hematological disorders, livedo reticularis, nephropathy, and valvular heart disease. 2 As presented, APS can also experience small vessel occlusions, leading to cutaneous ulcerations and digital gangrenes. These manifestations, although apparently less common, can severely impact patients’ outcome and should be therefore part of routine clinical assessment.
Finally, while the role of B-cells and immune dysregulation in APS pathogenesis provides a rationale for the use of Belimumab in this scenario, clear evidence of its beneficial effects is still lacking. In the presented case, we believe that the combined use of different therapeutic agents not only directed to counterbalance the procoagulant state, such as Belimumab and steroids, but also played a key role in achieving quick and full recovery.
To conclude, APS is a rare condition and despite the significant improvements that have been made in this field, it might still represent a diagnostic and therapeutic challenge for the treating clinicians. Rare and low-prevalence diseases are often underdiagnosed and misdiagnosed with negative consequences on patient’s quality of life and the risk of long-term complications. Indeed, the presence of specialized clinical centers aims to make timely diagnosis and choose the best therapeutic approach.
This clinical case highlights the importance of suspecting APS in patients experiencing unprovoked thrombosis, especially when at low risk for conventional cardiovascular risk factors. We have also emphasized the critical issue of testing aPL during anticoagulant treatment and focused on the need of aPL retesting in patients with high-titer positivity. One of the most relevant challenges in APS setting is represented by the identification of those clinical and serological characteristics which are associated with an increased risk of thrombotic recurrences and unusual manifestations of the disease, and in which a more aggressive and alternative treatment approach should be considered.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.