
Abstract
Objective
Systemic lupus erythematosus (SLE) is a multifactorial autoimmune disease in which dysregulated nucleic acid sensing and type I interferon responses drive autoantibody production and organ damage. Toll-like receptor 9 (TLR9) regulates these pathways, and promoter variants (rs187084 and rs5743836) may alter TLR9 expression and modulate disease susceptibility and severity. This study evaluated the association of these promoter SNPs and their haplotypes with SLE risk and clinical/laboratory features in an Iranian population.
Methods
In this case–control study, we genotyped TLR9 promoter SNPs rs187084 and rs5743836 in 140 SLE patients and 140 age- and sex-matched healthy controls using the real-time PCR-high resolution melting (PCR-HRM) assay.
Results
Our analysis showed that the CC and CT genotypes and the C allele of the rs5743836 SNP were associated with an increased risk of SLE (P < 0.05). However, there was no association between the rs187084 SNP and the risk of SLE (P > 0.05). The combined rs187084–rs5743836 CC haplotype was associated with increased SLE risk (CC vs TT haplotype, P < 0.001). Furthermore, in the patient group, carriers of the C allele in both promoter variants exhibited earlier disease onset and more active and severe disease, as evidenced by higher levels of C-reactive protein (CRP) and anti-dsDNA, reduced complement C3 and C4, and a higher frequency of renal and neurological complications (P < 0.05).
Conclusion
Our data suggest that promoter variation in TLR9, specifically the rs5743836 C allele and the combined rs187084-rs5743836 CC haplotype, is linked to an increased risk of SLE and more disease activity and severe clinical presentation.
Introduction
Systemic lupus erythematosus (SLE) is a chronic, relapsing autoimmune disorder characterized by a wide spectrum of clinical manifestations and variable outcomes, ranging from cutaneous and joint issues to life-threatening renal, neurologic, and hematologic involvement.1,2 The disease is fundamentally multifactorial: environmental exposures, hormonal effects, immune dysregulation, and complex genetic predisposition, which together shape individual risk, the patterns of organ involvement, the course of disease activity, and the extent of accumulated damage.3,4
Genetic factors significantly influence the susceptibility and characteristics of SLE. Family and twin studies have consistently shown a heritable component, with estimates of heritability ranging from 43% to 66% in different populations. The concordance rate for SLE in monozygotic twins is notably higher, ranging from 25% to 57%, compared to only 2% in dizygotic twins. 5 Genome-wide association studies (GWAS) have identified multiple susceptibility loci that implicate pathways related to innate and adaptive immunity, complement function, clearance of apoptotic debris, and lymphocyte signaling.6,7 While GWAS and other genetic studies have identified numerous loci linked to SLE, these genetic variants explain only a fraction of the heritability. These findings highlight the importance of regulatory variation and gene-environment interactions. Variants that influence gene expression or transcriptional regulation are likely to impact immune pathways crucial for the onset and development of the disease.8,9
Toll-like receptor 9 (TLR9) is a crucial innate immune sensor that detects unmethylated CpG motifs in microbial and endogenous DNA. It plays a significant role in linking nucleic acid detection to the MyD88-dependent type I interferon and proinflammatory cytokine production and activation of B cells.10,11 TLR9 signaling in plasmacytoid dendritic cells (pDCs) and B cells is involved in promoting interferon responses and the generation of autoantibodies, which are key processes in the development of SLE.12,13 In SLE and other autoantibody-mediated diseases, the defective clearance of apoptotic debris and the formation of nucleic acid-protein complexes facilitate the delivery of self-DNA to endosomes, which creates feed-forward loops of interferon production, B cell activation, and pathogenic immune complex formation. These loops drive tissue inflammation and organ damage.14,15
Multiple studies have shown an increase in TLR9 expression in cell types from patients with various autoimmune diseases, including rheumatoid arthritis, 16 Sjögren’s syndrome, 17 autoimmune thyroid diseases, 18 multiple sclerosis, 19 systemic sclerosis, 20 and particularly in SLE.21,22 Numerous studies report higher TLR9 expression in peripheral blood mononuclear cells and CD20+ B cells from SLE patients versus healthy controls, with levels rising further in those with lupus nephritis and correlating positively with SLEDAI and Renal-SLEDAI scores and histologic activity index.21,23–26 Accumulating functional and in silico evidence suggests that promoter variants in TLR9, specifically rs187084 (−1486T/C) and rs5743836 (−1237T/C), can impact transcription factor binding, promoter activity, and cytokine responses. These effects may play a role in autoimmune processes. These variants are located near NF-κB/ERα regulatory motifs. They create potential c-Rel/NF-κB, Sp-1, and IL-6 responsive sites, making them strong candidates for influencing susceptibility to SLE and its clinical manifestations.27–30
Given these findings, we investigated the association of rs187084 and rs5743836 with SLE susceptibility and key clinical and laboratory features of the disease in the Iranian population. By integrating genetic association analyses with clinical phenotype correlations, we aim to determine if functional promoter variation in TLR9 contributes to SLE risk or disease heterogeneity, with the potential to identify genetic markers indicative of TLR9-driven immune dysregulation.
Materials and methods
Samples
A total of 280 individuals were enrolled in this case–control study, comprising 140 patients with SLE and 140 healthy controls. Cases and controls were matched by age and sex and were drawn from the same population; all participants shared common ancestry and were unrelated. SLE patients were recruited from both inpatient and outpatient rheumatology clinics at Alzahra Hospital in Isfahan and met the 2019 EULAR/ACR classification criteria for SLE. Control subjects were healthy blood donors with no personal or family history of autoimmune or autoinflammatory and immunological diseases. Demographic and clinical information was obtained using a written questionnaire that recorded age, sex, age at disease onset, body mass index, family history of SLE and related disorders, and clinical manifestations. Routine laboratory assessments included serum complement components C3 and C4, anti-double-stranded DNA (anti-dsDNA) antibodies, erythrocyte sedimentation rate (ESR), white blood cell count, and C-reactive protein (CRP). Peripheral blood (5 mL) was collected into EDTA tubes from each participant after obtaining written informed consent and stored at −20°C until analysis. The study protocol and all procedures were approved by the local ethics committee of Isfahan University of Medical Sciences (IR.MUI.MED.REC.1404.168).
SNP genotyping
Genomic DNA was isolated from peripheral blood cells using the AddPrep Genomic DNA Extraction Kit (AddBio Inc., Korea) according to the supplied protocol. DNA yield and purity were assessed by UV–visible spectrophotometry, and integrity was checked by agarose gel electrophoresis. The genotyping of the SNPs was carried out utilizing the polymerase chain reaction high-resolution melting (PCR-HRM) method. The specific forward and reverse primers used to amplify fragments surrounding the variant sequences are: rs5743836 forward: CCTGCTTGCAGTTGACTGTG; rs5743836 reverse: CCCTGTTGAGAGGGTGACAT, and rs187084 forward: AGCTGACTGCTGGGTGTACATA; rs187084 reverse: TTTAACTGCTAGCACACCGGAT. The HRM assay was conducted using HOT FIREPol EvaGreen HRM Mix (without ROX) on a Rotor-Gene 6000 (Corbett Research). The protocol included an initial denaturation at 95°C for 10 min, followed by 40 cycles of 95°C for 20 s, annealing at 57°C for rs5743836 and 59°C for rs187084 for 25 s, and extension at 72°C for 20 s. Subsequently, melt data were collected from 60°C to 95°C with a ramp rate of 0.1°C/s and continuous fluorescence monitoring to generate high-resolution melting curves. Polymorphisms were identified by comparing the melting curve shapes of samples and normalized difference plots to reference genotypes previously confirmed by Sanger sequencing. Each run included three DNA samples with known genotypes as controls. To validate HRM results, 10% of the study samples were randomly chosen for confirmatory sequencing, and concordant results were used to confirm the assay performance.
Statistical analyses
All statistical analyses were conducted using IBM SPSS Statistics version 25 (Armonk, NY: IBM Corp). Continuous variables are reported as mean ± standard deviation, while categorical variables are presented as percentages. Genotype distributions in cases and controls were evaluated for deviation from Hardy–Weinberg equilibrium (HWE) using the chi-square (χ2) test. Differences in demographic and laboratory continuous variables between groups were assessed using the independent Student’s t-test. Comparisons of categorical variables, such as genotype and allele frequencies, were conducted using the Pearson chi-square test. Logistic regression was employed to assess the relationship between genotypes and case-control status, and to calculate odds ratios (ORs) with 95% confidence intervals (CIs) and corresponding P-values. Statistical significance was set at a two-sided P-value <0.05.
Results
Demographic, clinical, and laboratory profile
Baseline characteristics of SLE patients and control subjects who participated in the study.
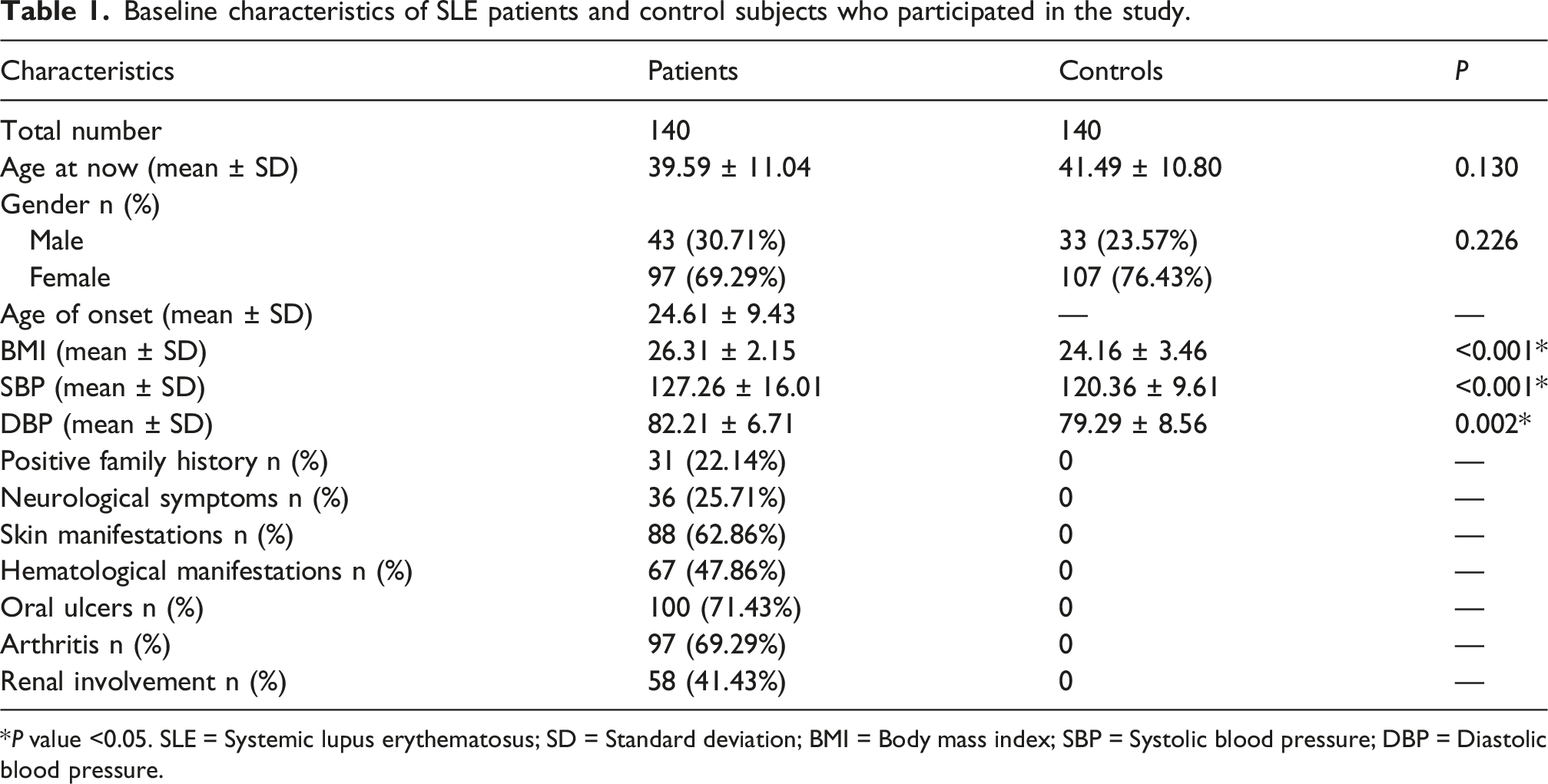
*P value <0.05. SLE = Systemic lupus erythematosus; SD = Standard deviation; BMI = Body mass index; SBP = Systolic blood pressure; DBP = Diastolic blood pressure.
Laboratory characteristics of patients with SLE and the control group.
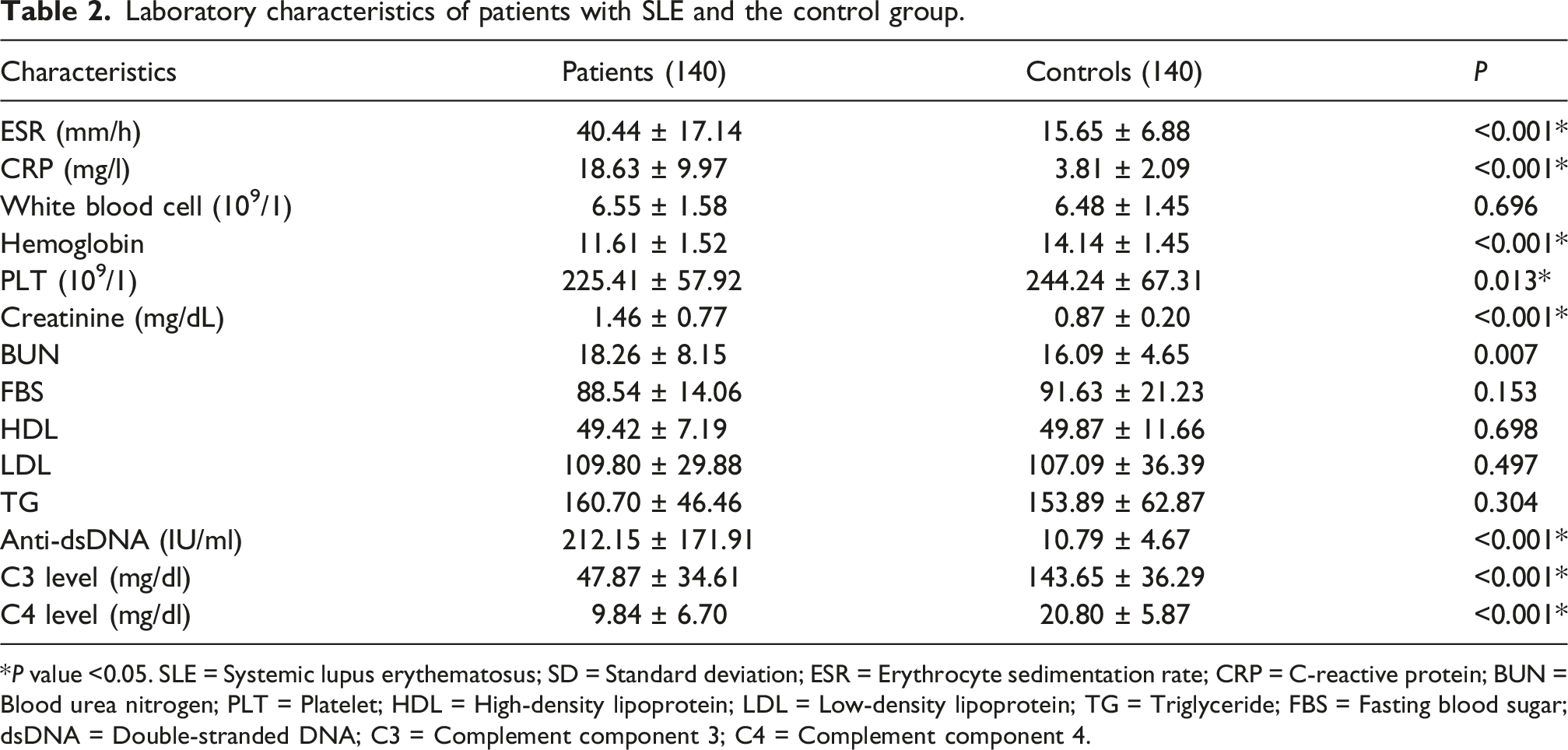
*P value <0.05. SLE = Systemic lupus erythematosus; SD = Standard deviation; ESR = Erythrocyte sedimentation rate; CRP = C-reactive protein; BUN = Blood urea nitrogen; PLT = Platelet; HDL = High-density lipoprotein; LDL = Low-density lipoprotein; TG = Triglyceride; FBS = Fasting blood sugar; dsDNA = Double-stranded DNA; C3 = Complement component 3; C4 = Complement component 4.
rs5743836 (−1237T/C): Frequencies and correlations
Association of TLR9 promoter polymorphisms with SLE risk.
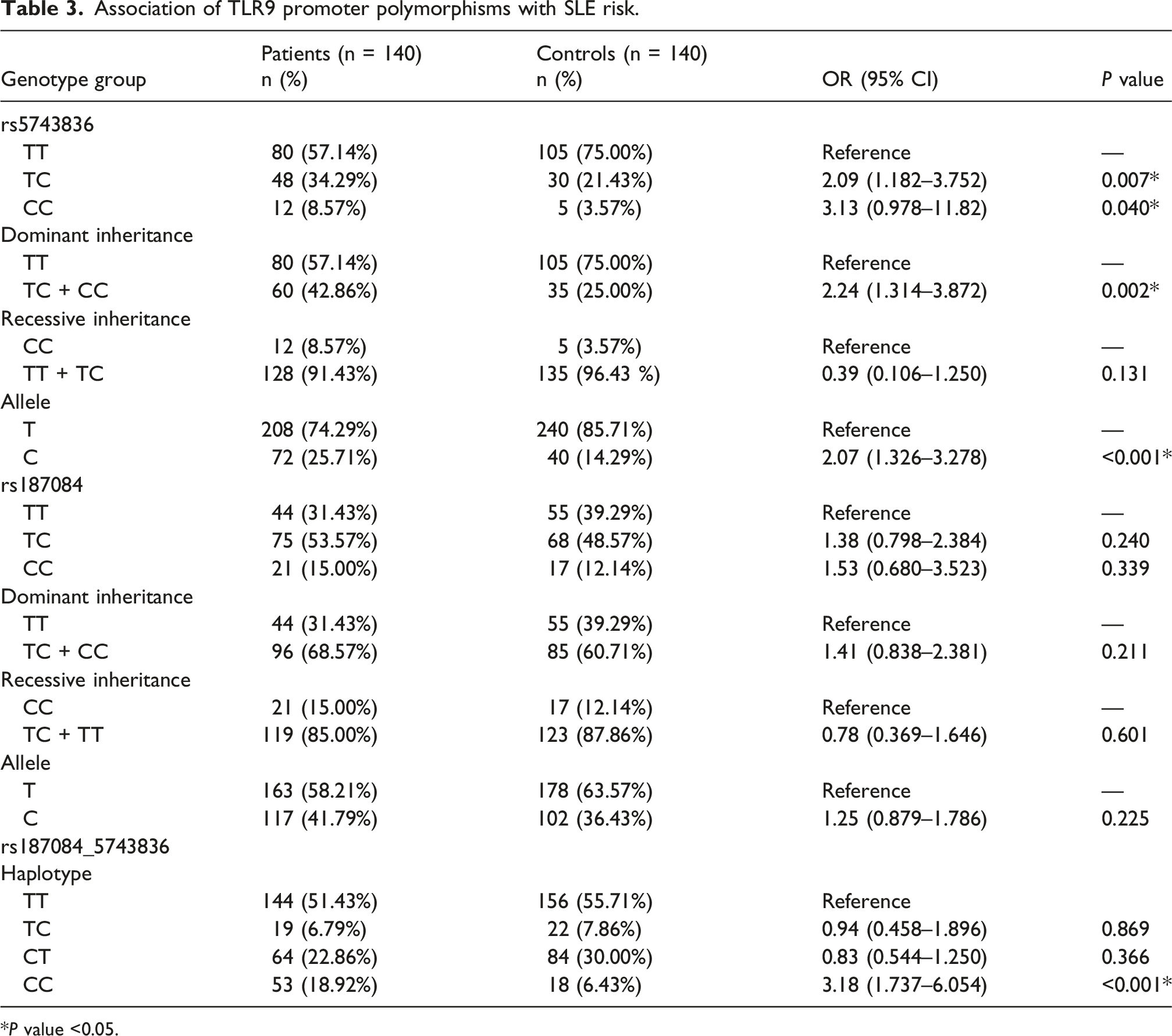
*P value <0.05.
Association of TLR9 (rs5743836) polymorphism with various parameters of SLE.
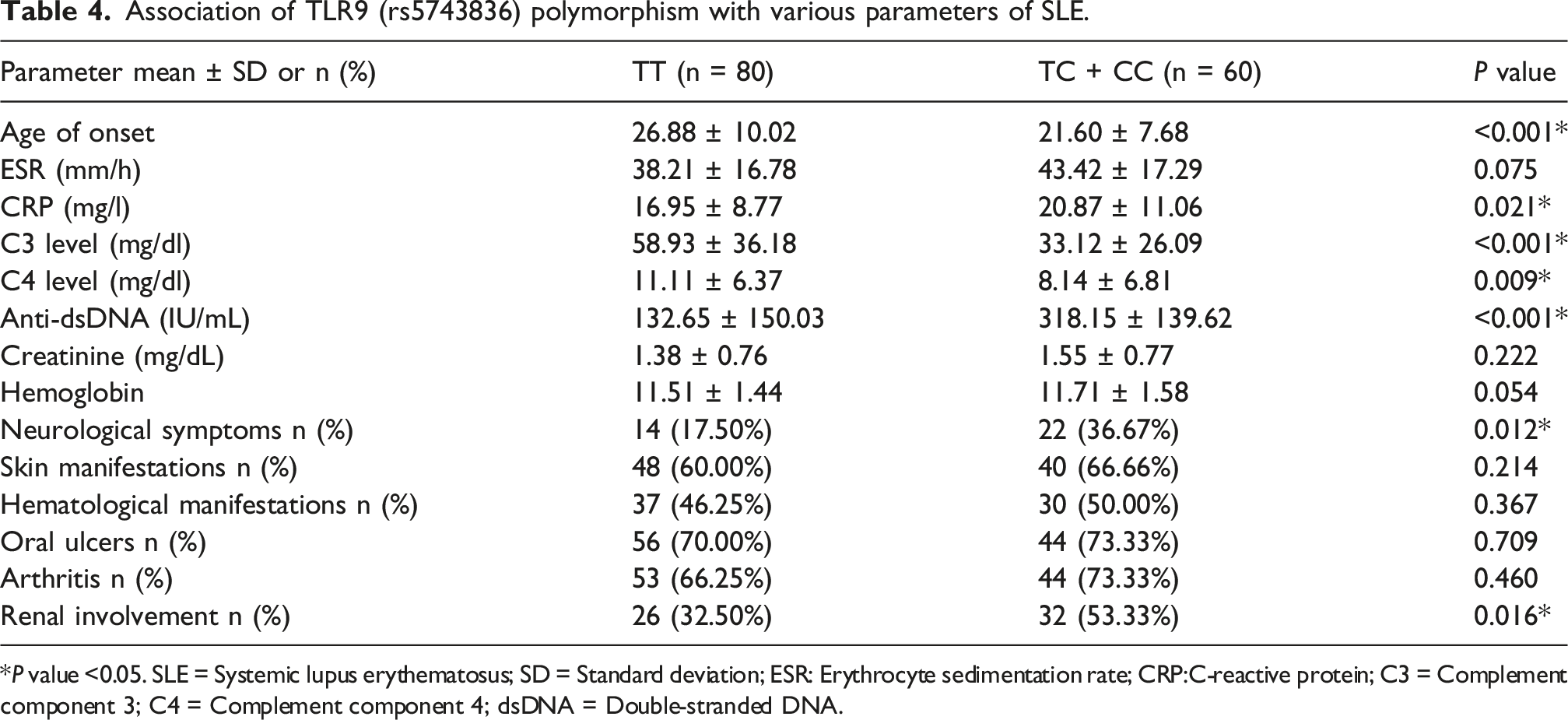
*P value <0.05. SLE = Systemic lupus erythematosus; SD = Standard deviation; ESR: Erythrocyte sedimentation rate; CRP:C-reactive protein; C3 = Complement component 3; C4 = Complement component 4; dsDNA = Double-stranded DNA.
rs187084 (‐1486T/C): Frequencies and correlations
The genotype frequencies in the case and control groups conformed to the Hardy-Weinberg equilibrium (HWE). The rs187084 genotype and allele distributions did not differ significantly between patients and controls. Genotype frequencies for TT, TC, and CC genotypes in SLE patients were 31.43%, 53.57%, and 15.00%, respectively, compared to 39.29%, 48.57%, and 12.14% in the control group. Our analysis of different inheritance models for the rs187084 polymorphism showed no increased or decreased risk for SLE under both dominant and recessive models (P > 0.05). The allele frequencies for T and C in SLE patients were 58.21% and 41.79%, respectively, while in non-SLE subjects, they were 63.57% and 36.43%. Overall, no statistically significant association with SLE was found for rs187084.
Association of TLR9 (rs187084) polymorphism with various parameters of SLE.
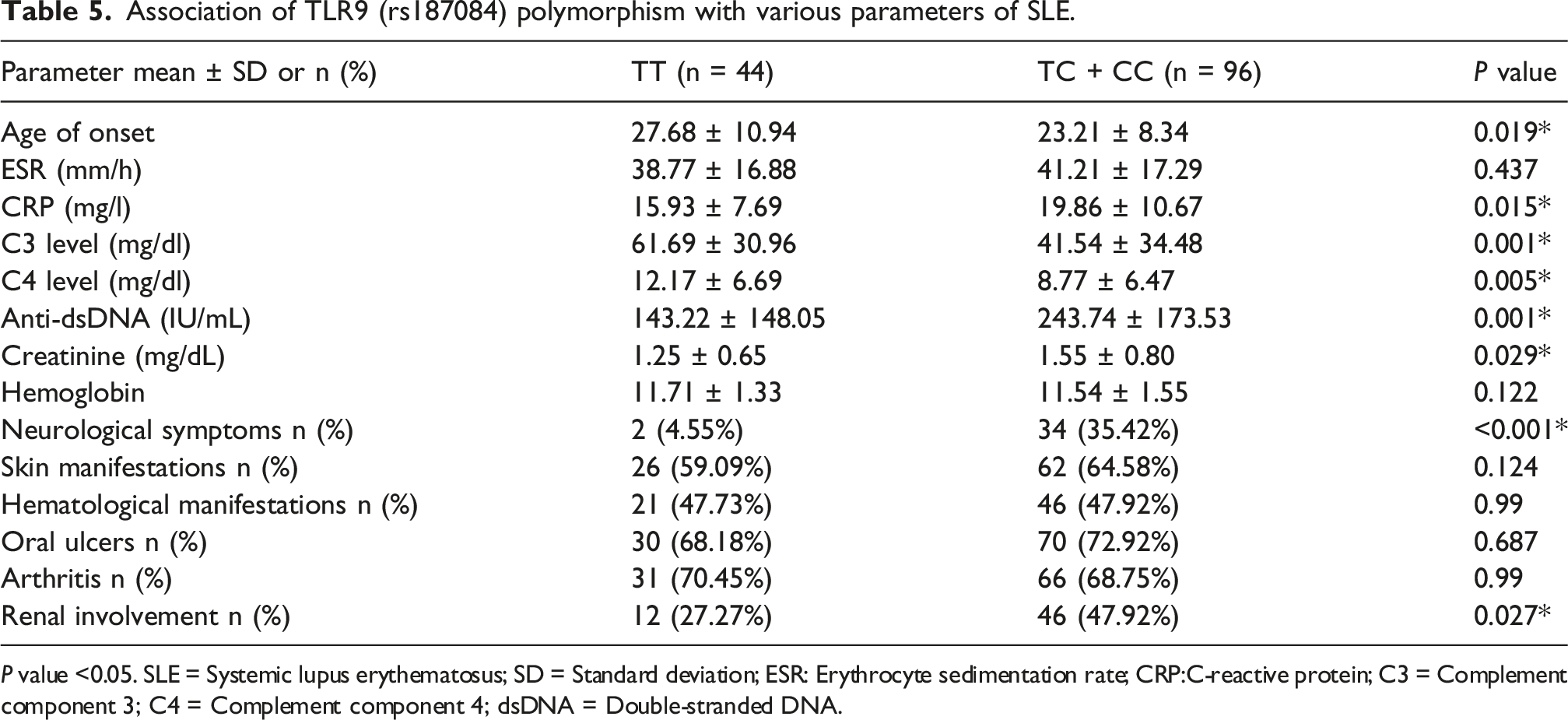
P value <0.05. SLE = Systemic lupus erythematosus; SD = Standard deviation; ESR: Erythrocyte sedimentation rate; CRP:C-reactive protein; C3 = Complement component 3; C4 = Complement component 4; dsDNA = Double-stranded DNA.
rs187084–rs5743836 haplotype association with SLE
The promoter haplotype at positions −1486T/C and −1237T/C showed that the CC haplotype was significantly more frequent in SLE patients (8.92%) than in controls (6.43%). Compared to the TT haplotype, the CC haplotype was associated with an increased risk of SLE (OR 3.18; 95% CI [1.74–6.05]; P < 0.001) (Table 3).
Discussion
Activation of TLR9 initiates MyD88-dependent signaling, leading to the activation of NF-κB and IRF transcription factors. This results in the production of proinflammatory cytokines such as TNF-α, IL-6, and type I interferons. In B cells, TLR9 signaling plays a role in activation, class switching, and differentiation into plasmablasts. Dysregulated TLR9 activity serves as a crucial link between nucleic acid sensing and the breakdown of tolerance, which is particularly relevant to the development of SLE.12,31,32 A wide range of experimental and clinical evidence suggests that TLR9 plays a significant role in the pathogenesis, disease activity, and organ-specific severity of SLE. Studies in lupus-prone mouse models have shown that genetic deletion of TLR9 greatly reduces or eliminates the production of spontaneous anti-dsDNA and antichromatin autoantibodies, indicating that TLR9 is essential for the development of anti-DNA autoreactivity. However, other autoreactive specificities, such as anti-RNA/Sm antibodies, persisted or even increased in the absence of TLR9. 33 In humans, multiple studies have reported increased TLR9 expression in various samples from SLE patients. Higher TLR9 levels are correlated with SLEDAI scores and markers of poor prognosis. Tissue analyses have shown increased TLR9 transcripts and protein in lupus nephritis biopsies, demonstrating robust proximal tubular TLR9 expression that parallels proteinuria and worsens glomerular injury. These observations implicate tubular TLR9 in the amplification of intrarenal inflammation and injury, suggesting that therapeutic strategies aimed at preventing or suppressing intrarenal TLR9 expression may mitigate renal damage in SLE.34,35 Clinically, persistently high TLR9 mRNA in whole blood is associated with poor prognosis and sustained disease activity over follow-up. Conversely, decreases in TLR9 expression are linked to favorable outcomes. These findings support a role for TLR9 as both a mediator of renal pathology and a potential biomarker of disease activity and severity in SLE. 36 Beyond SLE, TLR9 overexpression has also been observed in other autoimmune diseases, such as systemic sclerosis, specifically in CD3 + T lymphocyte cells. Notably, particularly high levels of TLR9 are found in the diffuse cutaneous subtype, correlating with skin fibrosis and increased disease severity. This suggests that TLR9 may play a role as a consistent amplifier of autoimmunity and tissue fibrotic processes, making it a potential marker for disease severity. 37
Given the central role of transcriptional regulation in TLR9 expression, promoter polymorphisms that alter transcription factor binding may influence TLR9 levels and, consequently, autoimmunity. Two functional variants in the TLR9 promoter, rs187084 and rs5743836, have been identified. In vitro promoter activation and luciferase reporter assays show increased activity for specific alleles of these SNPs compared to the wild-type sequence. Bioinformatic analyses indicate that the rs5743836 (C allele) creates or enhances binding motifs for transcription factors such as c-Rel/NF-κB, a site responsive to IL-6, and estrogen receptor α (ERα). In contrast, rs187084 is predicted to introduce or enhance a Sp1 binding site.27–29 Functional stimulation studies provide further evidence: PBMCs carrying the rs187084 C allele exhibit increased TLR9 transcriptional induction after microbial stimulation, 38 and PBMCs with the rs5743836 TC genotype show elevated TLR9 and IL-6 expression along with increased B cell proliferation in response to CpG, which depends on IL-6 signaling. 39 The proximity of rs5743836 to an NF-κB/ERα regulatory motif suggests a mechanism linking inflammatory and hormonal regulation of TLR9 expression. However, cell-type and allele-specific variability is observed; for example, some assays report higher activity with the T allele at position −1237. These findings underscore that the net effect of these SNPs depends on cellular context, stimuli, and regulatory factors. 30
Our study found that the rs5743836 C allele was associated with an increased risk of SLE and is consistent with prior reports of this allele’s association with upregulated TLR9 expression. Importantly, carriers of the C allele (TC + CC) in our cohort also presented with earlier disease onset and more active, severe disease, manifested by higher CRP and anti-dsDNA levels, reduced complement C3 and C4, and greater frequencies of renal and neurological complications. These results support the hypothesis that heightened TLR9 signaling contributes to elevated autoantibody production, inflammation, and organ damage in SLE. However, the literature on rs5743836 is heterogeneous. Santos et al. reported an increased SLE risk (OR = 1.59) and higher anti-SSa/Ro frequency for the C allele in a Southern Brazilian, primarily European-derived female sample. 40 Conversely, several large meta-analyses and population studies did not find a consistent association. Pooled analyses failed to show a risk for rs5743836 across Asian, European, Latin American, American, and African populations.41,42 Studies in Omani and some Caucasian (UK and US) cohorts likewise reported no overall SLE association.43–45 The Omani study linked rs5743836 to anti-cardiolipin IgM levels, a marker that in some reports correlates with disease activity.43,46,47 These discrepant results point to ancestry-specific effects, variable allele frequencies, and possible differences in linkage disequilibrium or environmental modifiers across populations.
Although rs187084 was not associated with overall SLE susceptibility in our cohort, carriers of the rs187084 C allele (TC + CC) exhibited phenotypes resembling those of rs5743836 C allele carriers: earlier age of onset, greater disease activity, increased frequency of renal involvement (including higher creatinine), and more neurological presentations. These findings support the hypothesis that the rs187084 C allele may modulate TLR9 expression or signaling, contributing to disease severity even if it does not independently increase population-level risk. This view aligns with a complex and partially inconsistent body of evidence, where ethnicity and study design influence observed associations. For example, a 2023 meta-analysis reported that the rs187084 T allele was associated with SLE in Asians but not in Arabs. 41 A Taiwanese study found the T allele linked to increased SLE risk but observed no genotype-phenotype correlations. 48 Other meta-analyses reported an overall association for rs187084 that disappeared upon ethnic stratification, and comprehensive reviews have failed to demonstrate a clear association across ancestries.42,49 Together, these data suggest that rs187084 may function more consistently as a modifier of clinical expression or via linkage with other regulatory variants in certain genetic backgrounds; accordingly, our findings demonstrate that the rs187084_rs5743836 CC haplotype correlates with increased risk of SLE.
Our primary focus was on SLE, but the functional implications of TLR9 promoter variants extend to broader human health due to the receptor’s universal role in innate immunity. TLR9 plays a crucial role in pathogen recognition by detecting unmethylated CpG motifs, especially in viruses and intracellular bacteria. 50 Polymorphisms like rs187084 that affect TLR9 expression levels may impact the efficiency of the initial immune response to various infectious challenges. Individuals with high-expression haplotypes may show enhanced clearance of certain pathogens, while those with lower-expression variants could be more susceptible to infections with CpG DNA-utilizing microbes. 51 Additionally, sustained, low-grade activation of this pathway, influenced by promoter variants, is linked to chronic inflammatory conditions beyond autoimmune diseases. The clinical impact observed in SLE likely represents an extreme manifestation of a genetic predisposition that subtly influences immune homeostasis, affecting general health and resistance to infectious diseases.
In conclusion, our data suggest that promoter variation in TLR9, specifically the rs5743836 C allele and the combined rs187084_rs5743836 CC haplotype, is linked to increased risk of SLE and more severe clinical presentation. This includes earlier onset, higher serologic activity, greater complement consumption, and increased renal and neurological involvement. These findings support the idea that heightened TLR9 signaling contributes to elevated autoantibody production, inflammation, and organ damage in SLE. Limitations of our study include a relatively small sample size, which may reduce the power to detect rare genotypes and result in wider confidence intervals. In addition, SLEDAI scores and some important clinical/laboratory manifestations, including nephritis and proteinuria, were not available for all patients due to incomplete clinical records. Therefore, associations between these parameters and TLR9 genotypes could not be comprehensively evaluated. Furthermore, the functional impact of these variants may vary among different ethnic groups due to genetic heterogeneity and environmental influences. Thus, these polymorphisms might reflect population-specific genetic patterns rather than direct causal roles in disease pathogenesis. Additionally, potential residual population stratification and the lack of direct functional or expression data linking the identified genotypes and haplotypes to changes in TLR9 transcription or downstream signaling are important considerations. To overcome these limitations, we recommend replication of our findings in larger, multiethnic cohorts, fine mapping, and evaluation of additional regulatory variants within and near the TLR9 gene. Integrated expression analyses, including mRNA and protein levels, should be conducted to establish genotype-expression correlations and assess the impact of different genotypes and haplotypes on TLR9 activity. This approach would clarify the causal relationship and potential clinical implications of TLR9 variants as prognostic biomarkers or therapeutic targets.
Footnotes
Acknowledgment
We would like to appreciate any support provided by Isfahan university of Medical Sciences.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.